Определение качества воды методами химического анализа.
Опыт № 5 Водородный показатель (рН)
Питьевая вода должна иметь нейтральную реакцию (рН около 7). Значение рН воды водоемов хозяйственного, питьевого, культурно-бытового назначения регламентируется в пределах 6,5 — 8,5.
Оценивать значение рН можно разными способами.
1. Приближенное значение рН определяется следующим образом.
В пробирку наливают 5 мл исследуемой воды, 0,1мл универсального индикатора, перемешивают и по окраске раствора определяют рН:
· Розово – оранжевая – рН около 5
2. Можно определить рН с помощью универсальной индикаторной бумаги, сравнить её окраску со шкалой.
3.Наиболее точно значение рН можно определить на рН – метре или шкале набора Алямовского.
По результатам нашего исследования:
Октябрьский район – рН около 6 — кислая
Ульбинский район – рН около 5- кислая
ВЫВОД: Повышенная кислотность в воде Ульбинского, Октябрьского районов и КШТ свидетельствует о плохом качестве исследуемой воды. Такая вода отрицательно влияет на организм человека, и может вызвать заболевания желудочно-кишечного тракта.
Опыт № 6 Определение хлоридов и сульфатов
Концентрация хлоридов в водоемах – источниках водоснабжения допускается до 350 мг/л.
Много хлоридов попадает в водоемы со сбросами хозяйственно- бытовых и промышленных сточных вод. Этот показатель весьма важен при оценке санитарного состояния водоема. Таблица №4
| Осадок или помутнение | Концентрация хлоридов, мг/л |
| Опалесценция или слабая муть | 1-10 |
| Сильная муть | 10-50 |
| Образуются хлопья, но осаждаются не сразу | 50-100 |
| Белый объемистый осадок | Более 100 |
Качественное определение хлоридов с приближенной количественной оценкой проводят следующим образом. В пробирку отбирают 5мл исследуемой воды и добавляют 3 капли 10 %-ного раствора нитрата серебра. Приблизительное содержание хлоридов определяют по осадку или помутнению (см таблицу).
Определение содержания хлоридов
Содержание хлоридов (х) в мг/л вычисляют по формуле
Где, 1,773 – масса хлорид ионов (мг), эквивалентная 1 мл точно 0,05 н. раствора нитрата серебра; V-объем раствора нитрата серебра, затраченного на титрование, мл.
Для расчета по опыту мы взяли 8мг/л (нитрат серебра)
Вывод: в воде КШТ –сильная муть, около 10-50 мг/л хлоридов; Ульбинский и Октябрьский районы – слабая муть, около 1-10мг/л;
Качественное определение сульфатов с приближенной количественной оценкой проводят так:
В пробирку вносят 10мл исследуемой воды, 0.5 мл соляной кислоты (1:5) и 2мл 5%-ного раствора хлорида бария, перемешивают. По характеру выпавшего осадка определяют ориентировочное содержание сульфатов: при отсутствии мути концентрация сульфат ионов менее 5мг/л; при слабой мути, появляющейся не сразу, а через несколько минут – 5-10мг/л; при слабой мути, появляющейся сразу, после добавления хлорида бария, -10-100мг/л; сильная, быстро оседающая муть свидетельствует о достаточно высоком содержании сульфат –ионов (более 100мг/л).
КШТ – ярко выраженная муть, 10-100мг/л; Ульбинский р-н – слабая муть, 5-10мг/л; Октябрьский район – слабая муть, образующаяся сразу после добавления хлорида бария,10-100мг/л;
ВЫВОД: Значительное превышение ПДК обнаружено в исследуемой воде Октябрьского района и КШТ, что может стать причиной некоторых сердечно-сосудистых заболеваний.
Опыт №7 Обнаружение фосфат — ионов.
Реагент: молибдат аммония (12,5г (NH4 )2 МоО4 растворить в дистиллированной Н2 О и профильтровать, объем довести дистиллированной водой до 1л); азотная кислота (1:2); хлорид олова.
К 5мл подкисленной пробы воды прибавляют 2,0мл молибдата аммония и по каплям(6капель) вводят раствор хлорида олова. Окраска раствора синяя при концентрации фосфат ионов более 10мг/л, голубая более 1мг/л, бледно-голубая -более 0,01мг/л.
ВЫВОД: В воде Ульбинского района и КШТ окраска раствора бледно-голубая, содержание фосфат- ионов – более 0,01мг/л, Октябрьский район окраска голубая- более 1 мг/л.
Опыт №8 Обнаружение нитрат – ионов.
Реагент: дифениламин (1г (С6 Н5 )2 NH растворить в 100мл H2 SO4 )
К 1мл пробы воды по каплям вводят реагент. Бледно- голубое окрашивание наблюдается при концентрации нитрат –ионов более 0,001мг/л, голубое –более 1мг/л, синее- более 100мг/л.
ВЫВОД: концентрация нитрат –ионов со всех трех водозаборов одинаковая, более 0,001мг/л
Качественное и количественное обнаружение катионов тяжелых металлов
Методы анализа: качественный анализ, включающий в себя дробный метод, разработанный Н.А Танаевым .Он открыл ряд новых, оригинальных реакций, позволяющих обнаруживать в растворе какой-либо определенный катион в присутствии большого числа других катионов, не прибегая к их предварительному осаждению. Количественный анализ, включающий атомно-эмиссионный метод, основанный на излучении атомных спектров вещества, возбуждаемых в горячих источниках света, а также сравнение и обобщение информации с литературными источниками.
Опыт №9 Обнаружение ионов свинца ( Pb 2+ )
Реагент: хромат калия (10г К2 СrO4 растворить в 90мл H2 O)
В пробирку помещают 5мл пробы воды, прибавляют 1мл раствора реагента. Если выпадает желтый осадок, содержание катионов свинца более 100мг/л; если наблюдается помутнение раствора, концентрация катионов свинца более 20 мл/л, а при опалесценции – 0,1 мг/л [6, c97-98]
ВЫВОД: Самое высокое содержание свинца в воде КШТ более 100мг/л осадок желтого цвета; октябрьский район-помутнение, более 20мг/л; Ульбинский район – опалесценция, 0,1мг/л.
Опыт №10 Обнаружение ионов кальция (Са 2+ )
Реагенты: оксалат аммония (17,5г (NH4 )2 С2 О4 растворить в воде и довести до 1л); уксусная кислота (120мл ледяной СН3 СООН довести дистиллированной водой до 1л).
В 5 мл пробы воды прибавляют 3мл уксусной кислоты, затем вводят 8мл реагента. Если выпадает белый осадок, то концентрация ионов кальция 100мг/л; если раствор мутный — концентрация ионов кальция более 1мг/л, при опалесценции – более0,01мг/л.[6, с128-129]
ВЫВОД: Самое высокое содержание ионов кальция в пробе с Октябрьского района 100мг/л, КШТ и Ульбинский район наблюдается помутнение раствора- концентрация ионов более 1мг/л
Опыт №11 Обнаружение ионов железа ( Fe 2+ )
В пробирку помещают 5мл исследуемой пробы воды, добавляют несколько капель K3 [Fe(CN)6 ] красная кровяная соль. Окраска раствора приобретает цвет под названием: турбулинская синь[6, c194-195]
ВЫВОД: Самое высокое содержание ионов железа 2 содержится в воде с КШТ, т.к по яркости окраски на первом месте- вода с КШТ, на втором – Ульбинский район, на третьем- Октябрьский район.
Опыт №12 Обнаружение ионов железа ( Fe 3+)
В пробирку помещаем 5мл пробы воды, добавляют несколько капель К4 [Fe(CN)6 ] желтая кровяная соль. Окраска раствора приобретает цвет под названием: берлинская лазурь.
ВЫВОД: Самое большое содержание ионов железа3 в воде с Октябрьского района -яркий, насыщенный цвет, в остальных двух пробах окрас менее насыщенный.
Получив результаты эксперимента, мы обратились к альтернативе, т.е возможности замены водопроводной воды талой.
Молекула воды имеет угловое строение;[1]входящие в ее состав ядра образуют равнобедренный треугольник, в основании которого находятся два протона, а в вершине — ядро атома кислорода, межьядерные расстояния О—Н близки к 0,1 нм, расстояние между ядрами атомов водорода равно 0,15 нм. Из восьми электронов, составляющих внешний электронный слой атома кислорода в молекуле воды две электронные пары образуют ковалентные связи О—Н, а остальные четыре электрона представляют собой две неподеленных электронных пары.
Атом кислорода в молекуле воды находится в состоянии sp2-гибридизации. Поэтому валентный угол НОН (104,3°) близок к тетраэдрическому (109,5°). Электроны, образующие связи О—Н, смещены к более электроотрицательному атому кислорода. В результате атомы водорода приобретают эффективные положительные заряды, поскольку на них создаются два положительных полюса. Центры отрицательных зарядов неподеленных электронных пар атома кислорода, находящиеся на гибридных — орбиталях, смещены относительно ядра атома и в свою очередь создают два отрицательных полюса.
Молекулярная масса парообразной воды равна 18 ед. Но молекулярная масса жидкой воды, определяемая путем изучения ее растворов в других растворителях, оказывается более, высокой. Это происходит из-за того, что в жидкой воде происходит ассоциация отдельных молекул воды в более сложные агрегаты (кластеры). Такой вывод подтверждается и аномально высокими значениями температур плавления и кипения воды. Ассоциация молекул воды вызвана образованием между ними водородных связей. По своей структуре вода представляет собой иерархию правильных объемных структур, в основе которых лежит кристаллоподобные образования, состоящие из 57 молекул и взаимодействующие друг с другом за счет свободных водородных связей. Это приводит к появлению структур второго порядка в виде шестигранников, состоящих из 912 молекул воды.
Свойства кластеров зависят от того, в каком соотношении выступают на поверхность кислород и водород. Конфигурация элементов воды реагирует на любое внешнее воздействие и примеси, что объясняет чрезвычайно лабильный характер их взаимодействия. В обычной воде совокупность отдельных молекул воды и случайных ассоциатов составляет 60% (деструктурированная вода), а 40% — это кластеры (структурированная вода).
источник
КОЛИЧЕСТВЕННЫЙ ХИМИЧЕСКИЙ АНАЛИЗ ВОД
МЕТОДИКА ВЫПОЛНЕНИЯ ИЗМЕРЕНИЙ МАССОВОЙ КОНЦЕНТРАЦИИ ИОНОВ НИКЕЛЯ В ПРОБАХ ПИТЬЕВЫХ, ПРИРОДНЫХ И СТОЧНЫХ ВОД МЕТОДОМ ИНВЕРСИОННОЙ ВОЛЬТАМПЕРОМЕТРИИ
УТВЕРЖДАЮ
Заместитель Министра В.Ф.Костин 20 марта 1996 г.
Методика допущена для целей государственного экологического контроля.
Методика рассмотрена и одобрена Главным управлением аналитического контроля и метрологического обеспечения природоохранной деятельности (ГУАК) и Главным метрологом Минприроды РФ
Главный метролог Минприроды РФ 20.03.96 г.
Начальник ГУАК Г.М.Цветков 20.03.96 г.
Настоящий документ устанавливает методику количественного химического анализа проб природных, питьевых и сточных вод для определения в них ионов никеля при массовой концентрации никеля от 1 до 2500 мкг/дм . При определении содержания ионов никеля (II) в пробах вод концентрация органического углерода в электролизере электрохимической ячейки не должна превышать 10 мг/дм . Мешающее влияние органической составляющей вод при содержании органического углерода выше 10 мг/дм устраняется обработкой пробы ультрафиолетовым облучением. Мешающее влияние 100-кратного избытка ионов меди (II), 50-кратного избытка ионов кадмия (II) и 10-кратного избытка ионов (II) устраняют добавлением пиридина.
Нормы погрешности измерений массовой концентрации ионов никеля регламентированы ГОСТ 27384-87* «Вода. Нормы погрешности измерений показателей состава и свойств».
________________
* На территории Российской Федерации документ не действует. Действует ГОСТ 27384-2002. — Примечание изготовителя базы данных.
Методика количественного химического анализа обеспечивает с вероятностью 0,95 получение результатов анализа массовых концентраций ионов никеля с погрешностью, не превышающей значений, приведенных в таблице 1.
Значения характеристики погрешности измерений и ее составляющих
Диапазон анализируемых концентраций, мкг/дм
Наименование метрологической характеристики
Характеристика случайной составляющей погрешности, , % ( )
Характеристика систематической составляющей погрешности, , % ( )
4.1. Анализатор инверсионный вольтамперометрический по ТУ 4215-001-05828695-95* (НПВП «ИВА») в комплекте с компьютером типа IBM PC с процессором 80486 или выше (операционная система Windows 95/98, свободный последовательный порт RS 232).
________________
* ТУ, упомянутые здесь и далее по тексту, являются авторской разработкой. За дополнительной информацией обратитесь по ссылке. — Примечание изготовителя базы данных.
4.2. Ячейка электролитическая, в состав которой входят:
— стакан стеклянный вместимостью 50 см типа В-1-50ТС по ГОСТ 25336, который выполняет функции электролизера;
— вспомогательный электрод (стержень из стеклоуглерода диаметром 0,2-0,5 см или графита спектрально чистого диаметром 0,5-0,6 см);
— электрод индикаторный (рабочий) графитсодержащий: типа I (импрегнированный, ИГЭ) или типа IV (толстопленочный, ТГЭ) (НПВП «ИВА”).
— электрод сравнения — хлорсеребряный лабораторный насыщенный типа ЭВЛ-1МЗ по ГОСТ 17792.
4.4. Весы лабораторные аналитические общего назначения с наибольшим пределом взвешивания 200 г, 2-го класса точности по ГОСТ 24104*.
________________
* На территории Российской Федерации документ не действует. Действует ГОСТ Р 53228-2008. — Примечание изготовителя базы данных.
4.5. Колбы мерные наливные стеклянные 2-го класса точности по ГОСТ 1770-74 исполнения 1 или 2 вместимостью 1000 см , 100 см , 50 см и 25 см с притертыми пробками; цилиндры вместимостью 50 см и 25 см .
4.6. Пипетки мерные лабораторные стеклянные 2-го класса точности по ГОСТ 20292-74*, вместимостью 10 см исполнения 2 или 3, вместимостью 5 см исполнения 1, вместимостью 1 см исполнения 4 или 5.
________________
* На территории Российской Федерации документ не действует. Действуют ГОСТ 29169-91, ГОСТ 29227-91 — ГОСТ 29229-91, ГОСТ 29251-91 — ГОСТ 29253-91. — Примечание изготовителя базы данных.
4.7. Дозаторы типа ПЛ-01-20, ПЛ-01-200, ПЛ-01-100 или другие с дискретностью установки доз 1,0 или 2,0 мкл.
4.8. Аппарат для приготовления бидистиллированной воды (стеклянный) типа АСД-4 по ГОСТ 15150-69, ТУ 25-1173, 103-84.
4.9. Установка для обработки проб ультрафиолетовым облучением типа 705 UV-Digester («Metrohm», Швейцария).
4.10. рН-метр-милливольтметр типа pH-150.
4.11. Установка для фильтрования под вакуумом с приспособлением для создания вакуума.
5.1. Государственный стандартный образец (ГСО) состава водных растворов ионов никеля (II) с погрешностью не более 1% отн. при 0,95 с концентрацией 1 мг/см .
5.2. Никель хлористый ( ) по ТУ 6-09-02-331-80, ос.ч.
5.3. Калий (натрий) хлористый по ГОСТ 4234 (4233)-77, х.ч., или ТУ 6-09-3678 (3658)-74, ос.ч. и растворы 2 моль/дм и 0,2 моль/дм , приготовленные на тридистиллированной воде.
5.4. Кислота серная по ГОСТ 14262-78, ос.ч.
5.5. Кислота хлористоводородная по ГОСТ 14261-77, ос.ч. плотностью 1,19 г/см .
5.6. Кислота азотная по ГОСТ 11125-84, ос.ч. и раствор 1 моль/дм .
5.7. Этанол по ТУ 6-09-4512-77, ос.ч.
5.8. Диметилглиоксим по ГОСТ 5828-77, ч.д.а.
5.10. Аммиак водный 25% раствор по ГОСТ 24147-80, ос.ч.
5.11. Хлорид аммония по ГОСТ 3773-72, х.ч. или ТУ 6-09-587-75, ос.ч.
5.12. Калий марганцевокислый по ГОСТ 20490-75, х.ч.
5.13. Вода бидистиллированная по ТУ 6-09-2502-77.
5.14. Вода тридистиллированная.
Воду тридистиллированную готовят перегонкой бидистиллированной воды в стеклянном или кварцевом аппарате без резиновых соединений в присутствии серной кислоты и раствора калия марганцевокислого (2-3 см 5% раствора калия марганцевокислого и 0,5 см концентрированной серной кислоты на 1 дм бидистиллированной воды).
5.15. Фильтры обеззоленные (синяя лента).
5.16. Фильтры мембранные со средним диаметром пор 0,5 мкм. Диаметр диска 35-55 мм.
5.17. Универсальная индикаторная бумага.
Реактив по п.5.2 применяется при отсутствии ГСО.
Измерения массовой концентрации никеля выполняют методом, основанном на адсорбционном концентрировании на поверхности графитсодержащего электрода комплексного соединения никеля (II) с диметилглиоксимом. Максимальный катодный ток восстановления комплексного соединения, локализованного на поверхности рабочего электрода, прямо пропорционально зависит от содержания ионов (II) в растворе в интервале 1,0-200 мкг/дм (II) и является аналитическим сигналом (АС). Массовую концентрацию никеля в растворе определяют методом добавки аттестованного раствора ионов никеля (II).
7.1. При выполнении анализов необходимо соблюдать требования техники безопасности при работе с химическими реактивами по ГОСТ 12.4.019*.
________________
* На территории Российской Федерации документ не действует. Действует ГОСТ 12.4.103-83. — Примечание изготовителя базы данных.
7.2. Электробезопасность при работе с электроустановками по ГОСТ 12.1.019*.
________________
* На территории Российской Федерации документ не действует. Действует ГОСТ Р 12.1.019-2009. — Примечание изготовителя базы данных.
7.3. Организация обучения работающих безопасности труда по ГОСТ 12.04.004.
________________
* Вероятно ошибка оригинала. Следует читать: ГОСТ 12.4.004. — Примечание изготовителя базы данных.
7.4. Помещение лаборатории должно соответствовать требованиям пожарной безопасности по ГОСТ 12.1.004 и иметь средства пожаротушения по ГОСТ 12.4.009.
Выполнение измерений может производить химик-аналитик, владеющий техникой вольтамперометрического анализа и изучивший инструкцию по эксплуатации анализатора инверсионного вольтамперометрического.
Измерения проводятся в нормальных лабораторных условиях.
Температура окружающего воздуха 20±10 °С.
Атмосферное давление (97±10) кПа.
Относительная влажность (65±15)%.
Частота переменного тока (50±5) Гц.
Напряжение в сети (220±10) В.
10.1. Отбор и хранение проб воды.
10.1.1. Химическую посуду, применяемую в процессе анализа и для отбора проб, обезжиривают 10% водным раствором едкого натрия в течение 10-12 часов, промывают бидистиллированной водой, затем промывают раствором 1 моль/дм азотной кислоты и ополаскивают бидистиллированной водой. Затем посуду обрабатывают концентрированной серной кислотой, промывают тридистиллированной водой, заливают хлористоводородной кислотой квалификации ос.ч. разбавленной тридистиллированной водой в соотношении 1:100, выдерживают в течение 2-3-х часов, после чего вновь промывают тридистиллированной водой.
10.1.2. Пробы воды отбирают в полиэтиленовые бутыли, предварительно промытые отбираемой водой. Объем отбираемой пробы воды должен быть не менее 100 см .
10.1.3. Отобранные природные воды фильтруют через плотный фильтр (синяя лента) и подкисляют хлористоводородной кислотой квалификации ос.ч. до pH 2-3, добавляя 1 см концентрированной кислоты на объем пробы 1 дм . Фильтрование природных вод, содержащих небольшое количество мелкодисперсных взвешенных веществ, возможно проводить с использованием мембранных фильтров со средним диаметром пор 0,5 мкм под небольшим вакуумом. Сточные воды фильтруют через плотный фильтр (синяя лента) и измеряют значение pH пробы. Затем с помощью хлористоводородной кислоты или гидроксида натрия устанавливают pH пробы 2-3. Пробы выдерживают не менее 3-4-х часов перед выполнением измерений. Пробы, законсервированные таким образом, хранят в холодильнике при 4-6 °С не более 2-х недель. Незаконсервированные пробы анализируют в день отбора.
10.1.4. При отборе проб составляется сопроводительный документ по утвержденной форме, в котором указывается:
— цель анализа, предполагаемые загрязнители;
— место, время отбора;
— номер пробы;
— должность, фамилия, отбирающего пробу, дата.
10.2. Подготовка электрохимической ячейки к выполнению измерений.
Стеклянный стакан (электролизер) после проведения анализа обрабатывают концентрированной серной кислотой и промывают бидистиллированной водой. Электроды (индикаторный, вспомогательный, сравнения) промывают бидистиллированной водой. Затем электролизер и электроды (вспомогательный и сравнения) выдерживают в растворе хлористоводородной кислоты концентрации 0,1 моль/дм в течение 1-2-х минут и вновь промывают бидистиллированной водой.
10.3. Приготовление растворов, необходимых для выполнения измерений.
10.3.1. Приготовление основных растворов (ОР) никеля (II) с массовой концентрацией ионов никеля (II) 0,1 мг/см .
10.3.1.1. Приготовление основного раствора никеля (II) из государственного стандартного образца состава ионов никеля (II) с аттестованной концентрацией элемента 1 мг/см .
В мерную колбу вместимостью 50 см вводят 5 см стандартного образца состава никеля (II) и доводят объем раствора до метки бидистиллированной водой.
10.3.1.2. Приготовление основного раствора никеля (II) в отсутствии ГСО:
На аналитических весах взвешивают в химическом стакане 0,4049 г хлористого никеля и растворяют в бидистиллированной воде, содержащей 20 см концентрированной хлористоводородной кислоты. Раствор количественно переносят в мерную колбу вместимостью 1 дм . Объем раствора доводят до метки на колбе бидистиллированной водой.
Основные растворы устойчивы в течение 6 месяцев.
10.3.2. Приготовление аттестованных растворов никеля (II).
Ознакомиться с документом вы можете, заказав бесплатную демонстрацию систем «Кодекс» и «Техэксперт».
источник
Количественный химический анализ вод. Методика измерений химического потребления кислорода в пробах природных и сточных вод титриметрическим методом
Нормативный документ устанавливает методику измерений бихроматной окисляемости — химического потребления кислорода (ХПК) при обработке пробы воды ионом бихромата при определенных условиях для проб природных (поверхностных и подземных) и сточных (производственных, хозяйственно-бытовых, ливневых и очищенных) вод титриметрическим методом. Методика применима при содержании в воде органических веществ, эквивалентном потреблению кислорода в диапазоне от 4,0 до 2000 мг/дм3.
| Обозначение: | ПНД Ф 14.1:2:3.100-97 | ||||||||||||||||||||||||||||||||||
| Название рус.: | Количественный химический анализ вод. Методика измерений химического потребления кислорода в пробах природных и сточных вод титриметрическим методом | ||||||||||||||||||||||||||||||||||
| Статус: | взамен | ||||||||||||||||||||||||||||||||||
| Заменяет собой: | ПНД Ф 14.1:2.100-97 «Количественный химический анализ вод. Методика выполнения измерений химического потребления кислорода в пробах природных и очищенных сточных вод титриметрическим методом» | ||||||||||||||||||||||||||||||||||
| Дата актуализации текста: | 05.05.2017 | ||||||||||||||||||||||||||||||||||
| Дата добавления в базу: | 01.02.2017 | ||||||||||||||||||||||||||||||||||
| Дата введения в действие: | 01.12.2016 | ||||||||||||||||||||||||||||||||||
| Утвержден: | 01.09.2016 ФГБУ Федеральный центр анализа и оценки техногенного воздействия | ||||||||||||||||||||||||||||||||||
| Ссылки для скачивания: |
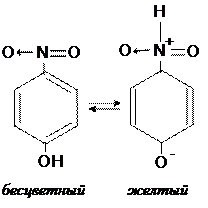 |
Рисунок 4.8 – Структура таутомерных форм индикаторного вещества
(п-нитрофенола), содержащего хромофорную и ауксохромную группы.
Хромофорная теория также имеет ряд недостатков, в частности:
Ø не объясняет, почему изменение окраски и таутомерные превращения зависят от значения рН среды;
Ø каким образом окраска большинства индикаторов, имеющих хромофорные группы, изменяется практически мгновенно, что противоречит механизму внутримолекулярной перегруппировки;
Ø и, наконец, хромофорная теория не поддается количественному описанию.
Ионно-хромофорная теория.
Эта теория объединила представления ионной (диссоциативной) и хромофорной теорий. Согласно ионно-хромофорной теории, кислотно-основные индикаторы – слабые кислоты и основания, причем нейтральные молекулы и их ионизированные формы содержат разные хромофорные группы. В водном растворе молекула индикатора способна либо отдавать ионы водорода (слабая кислота), либо принимать их (слабое основание), подвергаясь при этом таутомерным превращениям согласно схеме:
HInd Û H + + Ind — Û H + + Ind — B,
где HInd— неионизированная молекула индикатора (слабая кислота, таутомерная форма I) ; Ind — B— анион сильной кислоты, имеющей таутомерную форму II в диссоциированном состоянии (основная форма II).
При понижении рН (подкисление раствора) равновесие в системе смещается влево в сторону неионизированной формыHInd. Как только она начинает доминировать , раствор приобретает ее окраску.
Если раствор подщелачивать (рН увеличивается, а концентрация H + — убывает) – равновесие в системе смещается вправо и доминирующей формой становится Ind — B, которая и придает раствору иную окраску, характерную уже для основной формы II. Так кислая форма фенолфталеина (рН = 8,2) – бесцветна, а при переходе в щелочную среду – образуется анион таутомерной основной формы (рН = 10), окрашенный в красно-малиновый цвет. Между этими формами существует диапазон значений рН (от 8,2 до 10), соответствующих постепенному изменению окраски индикатора.
Глаз человека способен воспринимать окраску только одной из двух форм в смеси, при условии одинаковой интенсивности их цвета, если концентрация одной из этих форм примерно в 10 раз выше, чем второй.
Индикаторов.
1. Кислотно – основные индикаторы – это слабые органические кислоты или основания. Окраска индикаторов обратима и определяется значением рН среды. Интервал перехода рассчитывается через константу диссоциации:
DрНинд. = – logКа ± 1, где Ка – константа диссоциации индикатора.
Рассмотрим пример. Константа диссоциации индикатора ализаринового желтого Ка = 10 -11 . Определим интервал перехода индикатора DрНинд:
DрНинд. = – log (10 -11 )± 1 =11 ±1 Þ DрНинд [(11-1) ¸ (11+1)] = [10 ¸ 12].
Интервал перехода индикатора DрНинд = 10 ¸ 12.
2. Редокс – индикаторы – органические вещества, проявляющие свойства слабых окислителей или восстановителей. Могут быть как обратимыми (дифениламин), так и необратимыми, окраска которых разрушается (метиловый красный, метиловый оранжевый, они известны также как кислотно-основные индикаторы). Изменению окраски индикатора соответствует обратимая реакция: Ind + + ne Û Ind;гдеInd + — окисленная (Ox), а Ind — восстановленная (Red) формы индикатора, n— число электронов в данной полуреакции. Изменение редокс-потенциала (интервал перехода индикатора) рассчитывают по уравнению Нернста: DЕ = Е 0 ± 0,059/n,
где Е 0 — стандартный редокс-потенциал для индикатора; n – число электронов в полуреакции.
Например: Редокс-индикатор дифениламин имеет Е 0 = + 0,76 В и n = 2. Определим интервал его перехода.
Согласно формуле: DЕ= 0,76 ± 0,059/2 = 0,76 ± 0,0295 Þ DЕ=(0,76 –0,0295)¸ (0,76 + 0,295) = 0,73 ¸ 0,79 (В).
3. Металлохромные (металлоиндикаторы) — это органические красители (слабые кислоты), имеющие собственные хромофорные группы,и обратимо изменяющие свой цвет при образовании комплексной соли с катионами металлов. Используют их преимущественно в комплексонометрии, например, эриохром черный Т. Для этих индикаторов дополнительно должно выполняться условие: устойчивость комплекса титруемого вещества с титрантом выше, чем комплексов, образуемых им с индикатором в растворе. Интервал перехода вычисляют по формуле:
DрМе = – logКуст. ± 1, где Куст — константа устойчивости комплекса, образованного данным индикатором с титруемым веществом.
4. Осадительные индикаторы.Группа индикаторов незначительна по составу, так как окрашенный осадок должен формироваться в растворе сразу же после практически полного осаждения определяемого вещества (остаточная концентрация менее 10 –6 моль/дм 3 ), а таких веществ немного.
Интервал перехода индикатора определяют по значению произведения растворимости (ПР), образованного им осадка: Dр(ПР) = – logПР. ± 1.
Адсорбционные индикаторы -это органические вещества,проявляющие свойства слабых кислот или оснований, такие как эозин или флуоресцеин.
Механизм действия адсорбционного индикатора показан на схеме (рис. 4.9). Как видно из рисунка 4.9, появление окраски происходит в результате изменения состава ионов на поверхности дисперсной фазы (осадок или коллоидная частица) за счет процессов адсорбции или десорбции ионов индикатора. Это явление объясняется сменой знака электростатического заряда поверхности частиц осадка в ходе титрования. Причина ее в том, что в недотитрованном растворе поверхность осадка преимущественно сорбирует титруемые ионы, которые входят в его состав (осадок AgCl сорбирует неоттитрованные ионы Cl — ) и приобретает их заряд. В результате этого сорбция ионов индикатора становится невозможной.
Рисунок 4.9 – Схематическое изображение структуры сорбированного слоя на поверхности осадка AgCl, образующегося при титровании ионов Cl — раствором AgNO3.
а – до точки эквивалентности (поверхностью сорбируются ионы Cl — , а ионы индикатора Ind — остаются в растворе);
б – после точки эквивалентности (поверхность сорбирует ионы титранта Ag + , которые притягивают ионы индикатора Ind — ).
Как только будет достигнута точка эквивалентности, в растворе появится избыток противоположно заряженных ионов титранта, которые также начнут скапливаться вблизи поверхности осадка, притягивая из раствора ионы индикатора. Образующееся в результате этого вещество окрашивает поверхность осадка.
5. Специфические индикаторы – Относительно малочисленная группа индикаторов, так как в основе их применения специфические реакции с титруемом веществом. Такими свойствами обладает раствор крахмала по отношению к молекулам J2: образование соединения синего цвета.
Способы титрования.
Так как напрямую, реакцией с титрантом, можно анализировать далеко не любое вещество, особенно, если оно неустойчиво на воздухе, то для решения подобных задач было разработано несколько приемов (способов) проведения анализа. Они позволяют заменять неустойчивые, в данных условиях соединения, на эквивалентное количество более устойчивого, которое не подвергается гидролизу или окислению. Известны следующие основные способы проведения титриметрического анализа:
Ø обратное титрование или титрование по остатку;
Ø косвенное титрование или по замещению (по заместителю).
В таблице 4.1 показаны области применения различных способов в зависимости от вида титрования.
Таблица 4.1 – Применение различных видов и способов титрования.
| название метода | частное название метода; (рабочий раствор) | вещества, определяемые титрованием | ||
| прямым | обратным | косвенным | ||
| Протолито-метрия | Ацидиметрия (кислоты: HCl) | основания; соли, образован-ные сильным основанием и слабой кислотой | соли слабых оснований и сильных кислот; органические соединения | — |
| Алкалиметрия (щелочи: NaOH) | кислоты; соли, образован-ные слабым осно-ванием и сильной кислотой | — | — | |
| Редокси-метрия | Перманганато-метрия ( 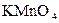 ) ) | восстановители | окислители | вещества, реагирующие с восстанови-телями |
Иодометрия (  и и  ) ) | восстановители | восстановители | окислители; кислоты | |
| Комплексо-метрия | Комплексоно- метрия (ЭДТА) | катионы, образующие с ЭДТА комплексы | катионы в водо-нерастворимых соединениях; катионы, для которых отсутствует индикатор | катионы, образующие с ЭДТА более устойчивый комплекс, чем с 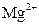 |
| Метод Седимен-тации | Аргентометрия (  ) ) | Анионы, образую-щие с  осадок осадок | катионы, образующие малорастворимый осадок с ионами галогенов:  , ,  , ,  ; ;  , ,  | — |
Рассмотрим подробнее суть различных способов титрования.
1. Прямое титрование заключается в непосредственном взаимодействии титранта и титруемого вещества. В процессе титрования к аликвоте или навеске вещества постепенно добавляют раствор титранта, объем которого точно фиксируют в Т. Э.В качестве титранта используют рабочий раствор известной концентрации. Расчет содержания вещества в образце выполняют по закону эквивалентов:
 =
=  (4.1)
(4.1)
где– количество моль-эквивалентов анализируемого вещества в титруемом образце; а  —количество моль-эквивалентов титранта, вступившего в реакцию с определяемым компонентом А.
—количество моль-эквивалентов титранта, вступившего в реакцию с определяемым компонентом А.
Концентрацию компонента А в растворе вычисляют по формуле:
 (4.2)
(4.2)
где 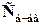 – молярная концентрация эквивалента (нормальность) титруемого раствора (определяемого компонента), моль-экв/л;
– молярная концентрация эквивалента (нормальность) титруемого раствора (определяемого компонента), моль-экв/л;  – объем аликвоты титруемого раствора, мл;
– объем аликвоты титруемого раствора, мл;  – концентрация и
– концентрация и  — объем титранта в точке эквивалентности. При титровании методом отдельных навесок формула (4.2) преобразуется в выражение (4.3):
— объем титранта в точке эквивалентности. При титровании методом отдельных навесок формула (4.2) преобразуется в выражение (4.3):
 (4.3)
(4.3)
Метод применяется во всех случаях, когда нет каких-либо ограничений. Например, при анализе кислот, определении жесткости воды.
2. Реверсивное титрование – это разновидность прямого титрования, когда рабочий и титруемый растворы меняют местами. В этом случае для анализаотбирают аликвоты рабочего раствора, а в Т.Э. измеряютизрасходованный натитрование объем анализируемого раствора. Вычисления проводят также, как и в прямом титровании, по формулам (4.2) или (4.3). Метод позволяет ограничить площадь поверхности раствора, контактирующей с воздухом, при стандартизации относительно неустойчивых соединений, как например NaOH.
Титрование по заместителю (косвенное) и титрование по остатку(обратное)основаны на использовании вспомогательного раствора, взаимодействующего с определяемым компонентом. Такой прием позволяет выполнять анализ химически нестойких объектов или же при отсутствии подходящего индикатора.
В косвенном титрованиисначала осуществляют реакцию определяемоговеществаАсо вспомогательным растворомВ,а затемтитруют эквивалентное количество образовавшегося продукта реакции С (заместитель). Этот способ можно представить в виде схемы: А + В  С + (т-т) , исходя из которой запишем выражение для закона эквивалентов:
С + (т-т) , исходя из которой запишем выражение для закона эквивалентов:
=  =
=  . (4.4)
. (4.4)
Из равенства (4.4) следует, что = и расчет можно также выполнять по формулам (4.2) и (4.3), используемых для прямого титрования. Для полноты реакции вспомогательный раствор всегда берут с небольшим избытком. Такой метод титрования реализуется в йодометрии.
Вобратном титровании также сначала протекает реакция между определяемымвеществомАивзятым в избыткевспомогательным растворомВ,но затем титруют остаток не прореагировавшего вспомогательного раствора. Поэтому необходимо точно знать концентрацию вспомогательного раствора В и его объем, взятый для анализа. Определение компонента А выполняется согласно схеме: А + В Вост + (т-т).Исходя из условий титрования, закон эквивалентов можно записать в виде:
 – = .(4.5)
– = .(4.5)
= — .(4.6)
Если все вещества взяты в виде растворов, то формула (4.6) примет вид
 (4.7)
(4.7)
Если хотя бы одно из веществ взято в сухом виде (известна его масса), то следует воспользоваться выражением (4.6) и записать значение  для каждого из веществ индивидуально.
для каждого из веществ индивидуально.
И способы их приготовления.
В титриметрии используют растворы, концентрация которых установлена каким-либо способом с высокой степенью точности. Такие растворы называют стандартными титрованными или просто титрованными. Растворы классифицируют по назначению и по способу установления их концентрации.
По назначению их условно делят на рабочие растворы и растворы стандартов (первичные и вторичные).
Рабочими называют растворы, которые используются непосредственно в анализе при определении содержания вещества. Если рабочий раствор не относится к стандартным, то его необходимо отстандартизировать непосредственно перед выполнением анализа, так как концентрация в процессе хранения могла существенно измениться. Точную концентрацию рабочего раствора находят путем титрования стандартного раствора или установочных веществ (метод точных навесок). Это касается, например, таких рабочих растворов, как: NaOH, Na2S2O3×5H2O.
Под стандартным раствором понимают такой титрованный раствор, который устойчиво сохраняет свою концентрацию при длительном хранении. Основное назначениестандартных растворов — определение точной концентрации рабочих и иных растворов, применяемых в титровании.
Процесс установления точной концентрации раствора путем его титрования по стандарту называется стандартизацией.
По способу определения концентрации различают первичные стандарты или растворы с приготовленным титром и стандартизированные растворы.
Стандартизированные растворы — это такие растворы, концентрация которых устанавливается по стандарту и заранее не может быть точно определена. К ним относятся растворы кислот, щелочей, гидролизующихся и гигроскопичных солей, а также веществ, которые могут реагировать с атмосферным кислородом и углекислотой. Известно множествоспособовприготовления стандартизированных растворов. Наиболее часто для этой цели применяют: приготовление по приближенной навеске (щелочи, соли), методы разбавления или смешения растворов (кислоты, соли), методы ионного обмена (растворы солей).
Стандартные растворыклассифицируютпо способу определения их концентрации.Различают: первичные стандарты или растворы с приготовленным титром и вторичные стандарты — растворы с установленным титром.
Первичные стандарты — это растворы, которые готовят либо по точной навеске вещества (рис. 4.10), либо путем разведения специально приготовленных стандартизированных реагентов – фиксаналов (рис. 4.11). Фиксанал представляет собой стеклянную запаянную ампулу, выпускаемую промышленностью и содержащую строго нормированное количество реагента, обычно рассчитанного на 1 л 0,1 н. раствора.
Приготовление раствора по точной навеске начинают с расчета ее массы по заданной концентрации (титру или нормальности) и объему колбы. Навеску стандартного вещества взвешивают на аналитических весах с точностью до 1×10 -4 г и количественно переносят в мерную колбу, где ее растворяют при перемешивании (рис. 4.10).
Рисунок 4.10 – Порядок операций при приготовлении раствора первичного
стандарта по точной навеске: 1 – мерная колба Мора; 2 – воронка;
3 – бюкс с навеской вещества; 4 – промывалка с дистиллированной водой;
5 – пипетка или капельница.
а – перенос навески вещества в мерную колбу; б – ополаскивание воронки;
в – доведение объема раствора стандарта до метки.
Этим методом обычно готовят растворы солей, таких как бура (Na2B4O7×10H2O), K2Cr2O7. Количество вещества в растворе находят или по значению точно взятой массы навески (при ее переносе необходимо тщательно промыть бюкс), или рассчитывают методом разности, определяя точную массу бюкса сначала с навеской, а затем – пустого, уже после переноса вещества в колбу. В случае необходимости — концентрацию раствора заново пересчитывают с учетом фактически взятой массы навески.
Порядок приготовления раствора методом разведения из фиксанала показан на рисунке 4.11. Чтобы стандарт, полученный этим методом, был качественным и отвечал всем требованиям, необходимо исключить потери вещества при вскрытии ампулы и переносе его в колбу, а также следить, чтобы осколки ампулы не попали в раствор. Это во многом зависит от правильности обращения с ампулой.
Рисунок 4.11 – Способ приготовления растворов первичного стандарта
методом разведения из фиксанала: 1 – мерная колба Мора на 1л;
2 – нижний боек; 3 – воронка; 4 – ампула фиксанала; 5 – верхний боек.
Перед использованием, ампулу следует ополоснуть дистиллированной водой и только затем ее вскрывать специальным бойком. Сразу же после переноса вещества в колбу, нужно тщательно промыть ампулу дистиллированной водой, не менее, чем 6-ти кратным ее объемом. Этот метод приготовления первичного стандарта проще, чем по точным навескам, но уступает ему в точности. Его используют не только для получения растворов солей, но и различных кислот.
Так как для приготовления раствора первичного стандарта пригодны только точная мерная посуда и аналитические весы, то и к веществам, применяемым для этой цели, предъявляют ряд обязательных требований. В качестве первичного стандарта можно использовать только такие реактивы, которые характеризуются:
Ø высокой чистотой (обычно не хуже, чем 99,99 – 99,999% — квалификации ч.д.а. и о.с.ч.);
Ø точным соответствием формульному составу и относительно высокой молекулярной массой;
Ø устойчивостью при хранении как в твердом виде, так и в растворе (отсутствие процессов гидратации, гидролиза, окисления и карбонизации);
Ø простотой в приготовлении и хорошей растворимостью;
Ø необратимостью реакции при стандартизации, селективностью;
Ø возможностью точной фиксации Т. Э. каким-либо методом.
Вторичным стандартом называют такие стандартизированные растворы, которые устойчивы при хранении и могут быть использованы для стандартизации других растворов.
Вторичные стандарты готовят как растворы приблизительной концентрации любым известным методом, а перед употреблением — определяют их точную концентрацию путем стандартизациипо первичному стандарту. Поэтому при приготовлении вторичных стандартов не требуется высокая точность измерения массы вещества или объема раствора, как в случае первичных стандартов. Для этой цели вполне пригодны технохимические весы и неточная мерная посуда (цилиндры, мензурки, градуированные пробирки).
Примером раствора, обладающего свойствами вторичного стандарта, является соляная кислота. Ее разбавленные растворы могут храниться длительное время, до 1-го месяца и более, без заметного изменения концентрации. Бура, используемая в протолитометрии для стандартизации HCl, относится к первичным стандартам и готовится по точной навеске. Тогда, как рабочий раствор NaOH – свойствами стандарта не обладает вообще и его концентрацию приходится устанавливать заново при каждом использовании.
И их применение в анализе
Последнее изменение этой страницы: 2017-01-19; Нарушение авторского права страницы
источник