Сравнение методов анализа спектрометров на индуктивно-связанной плазме, масс-спектрометров и атомно-абсорбционных спектрометров
Оптическая спектрометрия на индуктивно-связанной плазме (ICP-OES) представляет собой привлекательную технологию, заставившую многих исследователей задуматься, будет ли разумнее приобрести ICP-OES или же продолжать придерживаться привычной технологии атомной абсорбции (AAS) (1). В последнее время в качестве стандартного средства анализа представляется новая и более дорогая технология, масс-спектрометрия на индуктивно-связанной плазме (ICP-MS) (2).
Технология ICP-MS изначально предлагает, хоть и по более высокой цене, преимущества ICP-OES и пределы детектирования атомно-абсорбционной спектрометрии на графитовом стержне (GF-AAS). Не смотря на знаменитое предсказание Фассела, «…что атомно-абсорбционная спектроскопия отомрет к 2000 году…», недорогие пламенные атомно-абсорбционные спектрометры всегда найдут применение в маленьких лабораториях с простыми запросами.
В данной статье вкратце описаны все три технологии и подчеркиваются важные критерии, по которым оценивается их применимость к Вашим аналитическим задачам. В Таблице 1 дан контрольный перечень типичных аналитических требований, который может помочь в оценке данных методов анализа.
Таблица 1. Контрольный перечень аналитических требований
Сколько образцов в неделю?
Типы образцов? (Металлы, горные породы, жидкие промышленные отходы, почвы, и т.д.)
Какой метод растворения можно применять?
Сколько и какие элементы должны быть определены?
Имеет ли значение наличие хлора (опция анализа дальнего УФ диапазона в некоторых ICP-OES спектрометрах?
Каковы диапазоны концентрации?
Какого объема образцы могут быть исследованы?
Какие еще опции/вспомогательные устройства принимаются во внимание? Почему?
Насколько для Вас важна изотопная информация?
Какую сумму Вы можете потратить на приобретение или лизинг прибора?
Какова стоимость владения и текущие затраты на методы, необходимые для покрытия предъявляемых требований?
Какой уровень подготовки Вашего оператора?
Многие люди с опытом использования ICP-OES считают ICP-MS плазмой с масс-спектрометром в качестве детектора. Масс-спектроскописты предпочитают описывать ICP-MS как масс спектрометрию с плазменным источником. В любом случае, этот метод способен предоставлять изотопную информацию. Данная информация может помочь в преодолении многих проблем «спектральной» интерференции, имеющих место в масс спектрометре.
В общих чертах, система ввода образцов и плазма в спектрометрах ICP-OES и ICP-MS выглядят схоже. В технологии ICP-OES оптический спектр с типичным диапазоном 165-800 нм просматривается и измеряется, последовательно либо синхронно. Синхронный ICP-OES спектрометр может быстрее определять большое количество элементов, но является более дорогим, чем последовательный ICP-OES спектрометр. Это сильно зависит от количества элементов и необходимых концентраций. С недавнего времени некоторые ICP-OES спектрометры могут достигать до 120 нм (3), тем самым позволяя детектировать Cl на первичной длине волны 134,664 нм с sub-ppm пределами детектирования.
Спектрометр ICP-MS извлекает ионы, произведенные в плазме, в интерфейс, состоящий из конуса образца и конуса skimmer (отборника). Эта конфигурация позволяет дифференциально снизить давление от атмосферного давления до окончательного давления между 10-5 и 10-7 торр. Ионы, прошедшие через интерфейс, направляются через ионную оптику, которая оптимизирует траектории ионов, устраняя нейтральные образцы и свет, обычно с помощью фотонных ловушек. Затем ионы проходят сквозь масс фильтр, как правило, квадрупольную линзу, прежде чем отобранные ионы достигнут детектора.
Спектрометр ICP-MS предоставляет информацию для каждой атомной единицы массы (amu), или в дальтонах. Отношение массы иона к его заряду отображается и помечается m/z, в диапазоне массы 3-250 дальтон. Изотопную информацию можно использовать несколькими способами, включая измерения изотопных соотношений, часто используемые для Pb и U, которые не имеют постоянно присущего относительного содержания, и анализ образцов с неприсущими изотопными содержаниями.
Изотопное разбавление представляет собой метод ввода образцов с известной концентрацией чистого изотопа для очень точного определения концентрации элемента. Предварительным условием этой технологии является то, что анализируемый элемент должен иметь больше чем один изотоп.
Рисунок 1. Типичный квадрупольный ICP-MS спектрометр
Рисунок 2. Типичный последовательный ICP-OES (монохроматор) спектрометр
Рисунок 3. Типичный двухлучевой AAS спектрометр
Пределы детектирования ICP-MS спектрометров очень впечатляют (Таблица 3, стр. 10). Большинство пределов детектирования растворов находятся в диапазоне 1-10 частей на триллион (ppt). Этот показатель так же хорош, или лучше, чем у спектрометров GF-AAS, для большинства элементов в чистой воде, а также он покрывает намного большее количество элементов. ICP-OES спектрометры обычно характеризуются на два-три порядка величины худшими пределами детектирования по сравнению с ICP-MS, с большинством элементов в диапазоне 1-10 частей на биллион (ppb).
В последнее время появился ICP-OES спектрометр (11) с впечатляющими пределами детектирования в sub-ppb диапазоне для некоторых элементов, при использовании высокоразрешающего монохроматора с радиально наблюдаемой плазмой. Другие спектрометры смогли несколько улучшить свои показатели при использовании аксиально отображаемой индуцированной плазмы, хотя такому отображению свойственны проблематичные матричные эффекты.
Следует, однако, заметить, что выше приведенный комментарий о пределах детектирования ICP-MS спектрометров относится к простым растворам с низкими уровнями содержания других растворенных веществ. По пределам детектирования, относящимся к концентрациям в сухих веществах, преимущество ICP-MS может существенно снизиться до 50 раз по причине худшей способности анализа растворенных в воде веществ. Некоторые распространенные более легкие элементы (напр., S, Ca, Fe, K и Se) имеют сильные интерференции в спектрометрах ICP-MS, что существенно ухудшает пределы детектирования.
Способность осуществления анализа в дальнем УФ диапазоне, присущая некоторым ICP-OES спектрометрам, открыла возможность для некоторых новых применений, в особенности анализа присутствия Cl в маслах. В современных ICP-MS спектрометрах сейчас часто ликвидируют возможность работы с отрицательными ионами с целью снижения стоимости производства. Это привело к тому, что ICP-OES спектрометры остались единственным средством атомной спектроскопии, способным определять Cl, Br, и I.
Пламенный атомно-абсорбционный спектрометр обычно характеризуется худшими пределами детектирования, чем ICP-OES, за исключением анализа щелочных металлов, напр., Na, K.
Три метода анализа демонстрируют различные типы и сложность интерференционных проблем. По этой причине мы рассмотрим каждый метод в отдельности.
Спектральные интерференции в ICP-MS спектрометрах предсказуемы и их число меньше 300. Полиатомные и изобарные интерференции имеют место, когда изотоп имеет схожую массу с аналитом, вследствие чего разрешение спектрометра (обычно около 0,8 дальтон) не позволяет разрешить его, напр., 58 Ni на 58 Fe, 40 Ar на 40 Ca, 40 Ar 16 O на 56 Fe, или 40 Ar- 40 Ar на 80 Se.
Можно использовать элементные уравнения (схожие по принципу с внутри-элементной коррекцией в ICP-OES спектрометрах). Во многих случаях можно применять альтернативные изотопы с меньшим присущим относительным содержанием. Использование смешанных газов (небольшой процент других газов, таких как азот или аммиак, добавленный к основному газу аргону) иногда может быть эффективен в снижении интерференций.
В недавнее время метод соударения клеток предоставил ICP-MS спектроскопистам возможность измерения малых концентраций с использованием таких оптимальных масс. Однако критические комментарии в промышленности пока еще предостерегают от применения данного метода в проведении рутинных исследований. Оптимальный выбор газа все еще составляет проблему, особенно для лабораторий, осуществляющих повседневные повторяющиеся исследования.
Фон в ICP-MS спектрометре так низок, обычно 35 Cl 40 Ar на 75 As и 35 Cl 16 O на 51 V.
Избежание наличия HCl, HClO4, H3PO4 и H2SO4 в ICP-MS спектрометре является первостепенным (Таблица 5, стр. 11) в большинстве исследований. Если это не представляется возможным, можно использовать разделяющую хроматографию (микро-столбцы) до ввода образца в плазму. Многие предпочитают этот метод, так как он позволяет избавиться от нежелательных изотопов, а также дает возможность в то же время увеличить концентрацию. Другими методами, используемыми для преодоления этих проблем, являются: электро-термическое выпаривание (ETV) и использование смешанных газов. Еще одной очень дорогой альтернативой является высокоразрешающий ICP-MS спектрометр с магнитным сектором, который может разрешать массы меньше 0,1 дальтона. Это позволяет устранить многие спектральные интерференции. Опять же, метод соударения клеток может в будущем предложить преимущества в устранении таких интерференций на рутинной основе.
Растворы для ICP-MS анализа обычно готовятся в азотной кислоте, однако, иногда следует проявить осторожность (Таблица 4, стр. 10).
Любые ионы с двойным зарядом могут создавать спектральную интерференцию на половину m/z однозарядных ионов, напр., 138 Ba++ на 69 Ga+ или 208 Pb++ на 104 Ru+. Эти интерференции немногочисленны и могут быть существенно снижены или устранены путем оптимизации системы до начала анализа.
Эффекты переноса включают эффекты распылительной камеры (эффект «адаптации») и различия в вязкости между пробными растворами и калибровочными стандартами. Это приводит к изменению эффективности производства аэрозоля от раствора к раствору. Как правило, требуется подбор матрицы, хотя в качестве альтернативного метода может использоваться внутренняя стандартизация. Высокая скорость сканирования ICP-MS спектрометра действительно дает превосходные результаты при использовании внутреннего стандарта.
Эффекты ионизации могут создаваться образцами, содержащими высокие концентрации элементов Группы I и II. Могут понадобиться подбор матрицы, разбавление образца, стандартная примесь, разбавление изотопов, извлечение или отделение с помощью хроматографии. Подавители ионизации не могут использоваться из-за растворенных в воде веществ.
Эффекты пространственного заряда главным образом происходят позади skimmer конуса, где чистая плотность заряда становится сильно отличной от нуля. Высокая плотность ионов ведет к взаимодействию между ионами, присутствующими в ионном луче, создавая предпочтительную потерю световых ионов в присутствии тяжелых ионов, напр., Pb+ на Li+. Эти эффекты могут компенсироваться подбором матрицы или тщательным выбором внутренних стандартов вдоль диапазона массы аналитов, хотя это может быть затруднительно осуществить на практике. Разбавление изотопов будет эффективно, но дорого, а самым простым и наиболее эффективным методом является разбавление образца.
Спектральные интерференции в ICP-OES более многочисленны, и их сложнее устранить. В ICP-OES документировано более 50 000 спектральных линий, и матрица может создавать значительные проблемы, что вынуждает использовать высокоразрешающие спектрометры при анализе таких образцов, как металлы, химические вещества, горные породы. Межэлементная коррекция и спектральная очистка, широко применяемые в синхронной ICP-OES спектрометрии, дают лишь ограниченный положительный эффект по причине возрастания неопределенности.
Фон в ICP-OES может быть повышен или структурирован, что требует автономной коррекции фона. Сложная динамическая коррекция фона, если таковая имеется, очень полезна в улучшении точности анализа. Различные молекулярные соединения, такие как OH, дают пики или полосы, которые могут создавать аналитические проблемы при низких концентрациях аналитов, ухудшая пределы детектирования для реальных образцов.
Как и в ICP-MS, ICP-OES может использовать внутренние стандарты для преодоления эффектов матрицы, таких как эффекты «адаптации» распылительной камеры и разницы в вязкости между образцами и калибровочными стандартами.
Интерференцию от легко ионизируемых элементов можно минимизировать путем тщательного выбора условий каждого элемента или путем добавления подавителя ионизации, т.е. добавив избыток элемента Группы I.
В GF-AAS присутствует лишь незначительное количество спектральных интерференций, если применяется дейтериевая фоновая коррекция, напр., влияние Fe на Se, на 196,0 нм, но эти редкие интерференции можно исключить при использовании спектрометра GF-AAS Зеемана.
Для многих матриц требуется внимательное планирование этапа золообразования, чтобы минимизировать фоновый сигнал во время распыления.
Использование химических модификаторов может оказаться полезным в увеличении допустимой температуры золы. Например, химический модификатор Ni для определения Se позволяет поднять температуру золы до 1000 ºC до потери Se. Применение фоновой коррекции Зеемана может улучшить точность анализа по сравнению с фоновой коррекцией дугой D2, используемой во многих GF-AAS применениях.
Они могут вызываться распылением аналита в среду охлаждающего газа. Эти интерференции в последнее время были снижены с помощью конструкции изотермической трубки и использования платформ, задерживающих распыление аналита, вследствие чего образец распыляется в среду горячего инертного газа.
Эффекты матрицы проявляются изменяющимся удержанием аналита на графитовой трубке в зависимости от типа образца. Сухие и зольные стадии могут оказывать сильное влияние на форму переходного пика. Использование матричных модификаторов (напр., PdCl2) и горячее впрыскивание могут быть достаточно эффективными в снижении этих эффектов, а в некоторых случаях полезно использовать измерение пиковой зоны.
Из-за низкой температуры пламени воздух/ацетилен (2,200 ºC), присутствует много химических интерференций, например, PO4 на Ca и влияние драгоценных металлов на другие драгоценные металлы. Использование «освобождающих агентов» может устранить эти эффекты, напр., хлорид лантана для Ca в фосфатных растворах и оксид урана или оксид лантана для драгоценных металлов. Список очень длинный, поэтому методология включает много трудозатрат по улучшению точности до того, как рутинный анализ может быть принят. Во многих случаях эти интерференции хорошо документированы, но работа с опорными материалами может зачастую быть полезна для улучшения точности анализа.
Пламенная атомно-абсорбционная спектроскопия, как и ICP-MS и ICP-OES, использует распылитель и разбрызгиватель, таким образом, ей характерны похожие интерференции, такие как разницы в вязкости между образцами и калибровочными стандартами. Подбор матрицы зачастую является обязательным (из-за прямого всасывания образца), и часто используется метод стандартных добавок, особенно потому, что внутренний стандарт на современных атомно-абсорбционных спектрофотометрах невозможен. Эффектов «адаптации» распылительной камеры в пламенной AAS меньше из-за большого размера капли и объема аэрозоля в распылительной камере.
В большинстве применений пламя создает различный спектральный фон для различных образцов по сравнению с контрольными пробами и стандартами. По этой причине многие элементы определяются с использованием фоновой коррекции, что включает применение непрерывного источника D2.
В проведении рутинных исследований технология ICP-OES достигла такого уровня автоматизации, что относительно необученный персонал может использовать методы, созданные специалистом по ICP-OES, схожие по простоте применения с пламенной атомно-абсорбционной спектрофотометрией. До недавнего времени технология ICP-MS оставалась приоритетом профессионального химика, осуществляющего тонкую юстировку прибора перед проведением рутинного анализа. Тенденция к упрощению стала очевидна с 1993 года, и будет продолжаться в дальнейшем. Одной из причин является полностью компьютеризированное управление параметрами, сохраненными для определенного метода. Еще одной причиной является использование многозадачного графического пользовательского интерфейса, показывающего оператору несколько индикаторов достоверности данных на одном экране. Такое программное обеспечение также уменьшает время разработки методов анализа для по-прежнему сложного метода ICP-MS. Технология GF-AAS, хотя и относительно проста для рутинных исследований, требует значительных навыков в разработке методов анализа.
Современные ICP-OES спектрометры способны анализировать на рутинной основе до 10% TDS и даже до 30% для простых соляных растворов.
Хотя анализ 0,5% TDS в ICP-MS возможен в течение ограниченного времени, большинство химиков довольствуются 0,2% TDS максимум. Об этом следует помнить, если оригинальный образец является твердым веществом. Максимальный предел детектирования некоторых элементов в ICP-MS может быть не столь впечатляющим, когда выражается в твердом веществе, в сравнении с методом анализа ICP-OES.
Пламенная AAS обычно может обеспечить до 5% TDS, хотя эта цифра снижается до 1% для пламени N2O/C2H2.
GF-AAS спектрометры справляются с очень высокими уровнями растворенных в воде веществ.
Спектрометры ICP-MS могут иметь линейный динамический диапазон, превышающий 10 5 . Различные методы для расширения линейного диапазона до 10 7 включают десенсибилизацию одной из ионных линз, использование аналогового режима детектора, или использование отдельной чаши Фарадея в качестве второго детектора. Однако эти методы должны использоваться с осторожностью, так как высокие концентрации элементов матрицы могут создавать проблемы, лучшим решением которых является разбавление, и/или иметь различные характеристики кривой расширенного диапазона.
По этой причине, и из-за проблем с высокими уровнями растворенных в воде веществ, метод ICP-MS должен главным образом применяться для проведения анализа микропримесей/сверх-микропримесей. Исключение составляет использование изотопного разбавления. При применении технологии изотопного разведения были получены очень хорошие результаты с высокими концентрациями.
Пламенная AAS характеризуется линейным динамическим диапазоном приблизительно 10 3 , поэтому могут понадобиться постоянные разбавления различных элементов. По этой причине, разбавление сверх диапазона с использованием автоматического разбавителя является очень важным в большей части процесса пламенной AAS.
Спектрометр GF-AAS имеет очень ограниченный показатель LDR 10 2 -10 3 . Он может использоваться для более высоких концентраций, если наименее чувствительная линия присутствует и определена.
Анализ от определения микроэлементов до определения главного элемента можно осуществлять с помощью ICP-OES благодаря отличному линейному динамическому диапазону 10 6 . Спектрометр ICP-OES идеален для анализа до и включая процентные уровни при использовании радиального схемы наблюдения. По этой причине ICP-OES часто бывает необходим, чтобы удовлетворить нужды лаборатории, в дополнение к имеющимся ICP-MS или GF-AAS.
Кратковременная (один анализ) точность технологии ICP-MS обычно составляет 1-3%. Этот показатель в повседневных исследованиях улучшается благодаря использованию многочисленных внутренних стандартов. Более длительная точность (часы) все же составляет 5 (10 8 с расширенным диапазоном)
источник


- Откуда: Москва

Популярное сообщение!
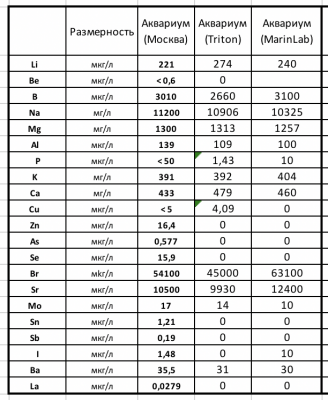
Все три пробы воды брались в один день и час

- Меня зовут: Юрий. Уже 60 !
- Откуда: Иваново
- Откуда: Москва
Что, в Москве работает ICP-лаборатория? По фосфору вообще шокирующая точность,

- Меня зовут: Леонид
- Откуда: Москва, Алексеевская

- Меня зовут: Дмитрий
- Откуда: Томск
Что, в Москве работает ICP-лаборатория? По фосфору вообще шокирующая точность,
- Меня зовут: Юрий. Уже 60 !
- Откуда: Иваново
если что там МКГ / Л. Почти в каждом городе есть такие лаборатории, у нас правда что ценник конский, что то около 4 тыс. за тест.
Точно! Сразу не въехал, что точность почти как у Салиферта. Ну, тадысь Ок!
- Откуда: Москва
А как ею воспользоваться? Если тут нельзя, то кино, пожалуйста, в личку.
У них нет коммерческих тестов. Надеюсь, пока.
- Откуда: Москва
если что там МКГ / Л. Почти в каждом городе есть такие лаборатории, у нас правда что ценник конский, что то около 4 тыс. за тест.
Да, примерно такая средняя цена за ICM тест по всем элементам.
- Меня зовут: Дмитрий
- Откуда: Томск
Да, примерно такая средняя цена за ICM тест по всем элементам.
интересно какая реальная себестоимость, понятно что оборудование очень дорогое, но интересно почему в европе тест дешевле стоит чем в у нас.

- Меня зовут: Надежда, Юрий
- Меня зовут: Дмитрий
- Откуда: Томск
Потому что все оборудование европейское или американское, покупается за доллары плюс таможня , плюс все препараты для тестов сново закупаются за рубежом , плюс снова таможня до 100 процентов.
логично, что то я об этом не подумал

- Меня зовут: Сергей
- Откуда: Калининград
интересно какая реальная себестоимость, понятно что оборудование очень дорогое, но интересно почему в европе тест дешевле стоит чем в у нас.
Marinlab и Triton являются ICP OES тестами, а в Москве делали ICP MS.
ICP MS это тот же OES плюс масс-спектроскопия, оборудование более дорогое, расходники требуются более чистые и специалисты совсем другого уровня.

- Меня зовут: Александр
- Откуда: Ухта, Республика Коми
А результат один. полная галиматья при сравнении трех. cм. пост #1

- Меня зовут: Андрей
- Откуда: Москва
Популярное сообщение!
Я исполнитель той самой колонки, что с подписью аквариум (Москва). У меня 15ти летний стаж в элементом анализе (ИСП ОЭС и МС), поэтому тоже позволю себе прокомментировать табличку.
Я заранее приношу извинения за множество профессиональных терминов, полностью избежать не удастся.
На мой взгляд результаты всех трех лаб очень даже удовлетворительные в плане сходимости.
Есть всего пара моментов в результатах зарубежных коллег, что меня смущают как специалиста.
Во первых нули.
Содержание элемента (именно элемента в морской воде) не может быть равно нулю в принципе. Здесь можно говорить о величине ниже которой методика (метод, прибор) не может «увидеть» присутствие элемента.
Эта величина называется предел обнаружения методики. Для каждого элемента (показателя, аналита) он свой. И расчитывается по результатам валидационного эксперимента. Это большая работа и она повышает себестоимость анализа, по крайней мере на начальном этапе.
С другой стороны подойдем
Предел обнаружения методики — это та величина выше которой мы уже можем говорить о точности! Точность выражается в неопределенности измерений. То есть грамотный аналитик обязан приписывать к своему результату процент неопределенности этой цифры.
И результат должен выглядеть так 100+-10 (сто плюс минус 10) Результат анализа это цифра определяющая центр доверительного интервала в котором лежит истинное значение. Внимание! с вероятностью 95%. То есть все равно остается 5% шанс что истинное значение не попадет в диапазон 90-110
Не сомневаюсь, что есть люди которые знают или как минимум интуитивно догадываются, что точность анализа это вероятность попадания в диапазон.
Однако большинство об этом не задумываются. Руководствуюсь верой, этому верю этому не верю.
Все эти моменты предпочитают не доносить до потребителя, и я поддерживаю такую тактику. Для непосвященного потребителя это лишний повод для сомнений.
Однако любой анализ имеет множество факторов вносящих неопределенность результата. Только одна флуктуация сигнала прибора указанная заводом изготовителем 1.5-3%, А ведь еще неопределенности измерений бланка, градуировки, раствор для коррекции и стандартов, разбавлений, человеческий фактор и тд. Все эти неопределенности суммируются (не впрямую математически). Поэтому конечная неопределенность менее 5% практически невозможна для любых сложных многофакторных и многостадиевых методов.
Капельные и индикаторные методы также не лучше в этом плане. Возьмите трех разных человек и пусть они несколько раз в разное время на разных партиях реактивов проведут анализ одного и того же раствора с добавкой. Полученные данные обработав статистически мы вряд ли получим неопределенность менее 5%. Один человек может получать хорошую сходимость за длительный период, но такие условия не отражают методику в целом, поскольку один человек может хорошо повторять случайную составляющую ошибки. Это не будет точность, это будет воспроизводимость.
Наверно совсем заморочил вам голову, но вообще давно хотелось этим поделится.
Бог с ней с неопределенностью, проще и выгоднее ее не писать. Но предел обнаружения, это тот пол на котором стоим. От которого начинаем видеть цифру не указывать считаю не верным.
У меня по фосфору ПО 50 мкг/л. Я увижу и ниже, может даже и 10 увижу, но не воспроизводимо! Неопределенность 10 мкг/л фосфора будет больше 100%.
Полтинника достаточно чтобы оценить концентрацию фосфатов (в пересчете на фосфат это кстати будет 150 мкг/л или 0.15 мг/л), зато честно, оперативно и комплексно.
Здесь же момент округления! Цифры с неопределенностью 10% бессмысленно выдавать с более чем двумя значащими знаками. Они уже не значимы.
Во вторых значения Фосфора и Иода
Вводят в смущение, поскольку значения очень низкие. Даже для современных приборов ИСП-ОЕС опуститься до фосфора в 1 мкг/л да же в чистой воде сомнительно, а то еще и в высокосолевой матрице морской воды.
Что же касается иода, то ИСП-ОЕС ваще с галогенами не очень дружат. 10 мкг/л у МаринЛаба весьма сомнительны для меня.
Я ни в коем случае не хочу очернить или уличить в некомпетентности своих зарубежных коллег. Они классные и хорошо что они есть. Еще раз повторюсь результаты всех трех лабораторий хорошо подтверждают друг друга!
За исключением может брома.
И опять же. Стронций у МаринЛаб больше на 25% чем у меня и у Тритона, это не значит что МаринЛаб ошибся. Это не стандартный образец с аттестованным содержанием стронция. Никто не знает его истинное значение. А для статистики трех результатов мало, минимум надо 15.
Я призываю оценивать цифру не буквально! (простите каламбур) А по ее значимости!
Получили цинк 250 мкг/л это сигнал — избыток цинка! и не важно 210 или 280, это все избыток! Не добавляйте цинк пока не спадет или ищите источник если он со временем не падает.
Получили железо 30 мкг/л — железо в норме! И не важно 20 или 50 — железо в норме

- Меня зовут: Александр
- Откуда: Вологодская область, город Череповец
Всем привет!
Я исполнитель той самой колонки, что с подписью аквариум (Москва). У меня 15ти летний стаж в элементом анализе (ИСП ОЭС и МС), поэтому тоже позволю себе прокомментировать табличку.
Я заранее приношу извинения за множество профессиональных терминов, полностью избежать не удастся.
На мой взгляд результаты всех трех лаб очень даже удовлетворительные в плане сходимости.
Есть всего пара моментов в результатах зарубежных коллег, что меня смущают как специалиста.
Во первых нули.
Содержание элемента (именно элемента в морской воде) не может быть равно нулю в принципе. Здесь можно говорить о величине ниже которой методика (метод, прибор) не может «увидеть» присутствие элемента.
Эта величина называется предел обнаружения методики. Для каждого элемента (показателя, аналита) он свой. И расчитывается по результатам валидационного эксперимента. Это большая работа и она повышает себестоимость анализа, по крайней мере на начальном этапе.
С другой стороны подойдем
Предел обнаружения методики — это та величина выше которой мы уже можем говорить о точности! Точность выражается в неопределенности измерений. То есть грамотный аналитик обязан приписывать к своему результату процент неопределенности этой цифры.
И результат должен выглядеть так 100+-10 (сто плюс минус 10) Результат анализа это цифра определяющая центр доверительного интервала в котором лежит истинное значение. Внимание! с вероятностью 95%. То есть все равно остается 5% шанс что истинное значение не попадет в диапазон 90-110
Не сомневаюсь, что есть люди которые знают или как минимум интуитивно догадываются, что точность анализа это вероятность попадания в диапазон.
Однако большинство об этом не задумываются. Руководствуюсь верой, этому верю этому не верю.
Все эти моменты предпочитают не доносить до потребителя, и я поддерживаю такую тактику. Для непосвященного потребителя это лишний повод для сомнений.
Однако любой анализ имеет множество факторов вносящих неопределенность результата. Только одна флуктуация сигнала прибора указанная заводом изготовителем 1.5-3%, А ведь еще неопределенности измерений бланка, градуировки, раствор для коррекции и стандартов, разбавлений, человеческий фактор и тд. Все эти неопределенности суммируются (не впрямую математически). Поэтому конечная неопределенность менее 5% практически невозможна для любых сложных многофакторных и многостадиевых методов.
Капельные и индикаторные методы также не лучше в этом плане. Возьмите трех разных человек и пусть они несколько раз в разное время на разных партиях реактивов проведут анализ одного и того же раствора с добавкой. Полученные данные обработав статистически мы вряд ли получим неопределенность менее 5%. Один человек может получать хорошую сходимость за длительный период, но такие условия не отражают методику в целом, поскольку один человек может хорошо повторять случайную составляющую ошибки. Это не будет точность, это будет воспроизводимость.
Наверно совсем заморочил вам голову, но вообще давно хотелось этим поделится.
Бог с ней с неопределенностью, проще и выгоднее ее не писать. Но предел обнаружения, это тот пол на котором стоим. От которого начинаем видеть цифру не указывать считаю не верным.
У меня по фосфору ПО 50 мкг/л. Я увижу и ниже, может даже и 10 увижу, но не воспроизводимо! Неопределенность 10 мкг/л фосфора будет больше 100%.
Полтинника достаточно чтобы оценить концентрацию фосфатов (в пересчете на фосфат это кстати будет 150 мкг/л или 0.15 мг/л), зато честно, оперативно и комплексно.
Здесь же момент округления! Цифры с неопределенностью 10% бессмысленно выдавать с более чем двумя значащими знаками. Они уже не значимы.
Во вторых значения Фосфора и Иода
Вводят в смущение, поскольку значения очень низкие. Даже для современных приборов ИСП-ОЕС опуститься до фосфора в 1 мкг/л да же в чистой воде сомнительно, а то еще и в высокосолевой матрице морской воды.
Что же касается иода, то ИСП-ОЕС ваще с галогенами не очень дружат. 10 мкг/л у МаринЛаба весьма сомнительны для меня.
Я ни в коем случае не хочу очернить или уличить в некомпетентности своих зарубежных коллег. Они классные и хорошо что они есть. Еще раз повторюсь результаты всех трех лабораторий хорошо подтверждают друг друга!
За исключением может брома.
И опять же. Стронций у МаринЛаб больше на 25% чем у меня и у Тритона, это не значит что МаринЛаб ошибся. Это не стандартный образец с аттестованным содержанием стронция. Никто не знает его истинное значение. А для статистики трех результатов мало, минимум надо 15.
Я призываю оценивать цифру не буквально! (простите каламбур) А по ее значимости!
Получили цинк 250 мкг/л это сигнал — избыток цинка! и не важно 210 или 280, это все избыток! Не добавляйте цинк пока не спадет или ищите источник если он со временем не падает.
Получили железо 30 мкг/л — железо в норме! И не важно 20 или 50 — железо в норме
Получили железо
- Меня зовут: Дмитрий
- Откуда: Томск
Я исполнитель той самой колонки, что с подписью аквариум (Москва). У меня 15ти летний стаж в элементом анализе (ИСП ОЭС и МС)
Доброго здравия вам и вашим питомцам!
Если не секрет это ведь наверное казенная установка ? у нас в городе просто таких прибора как минимум 4, в 3 можно заказать сумма везде правда одинаковая, хотя загруженность у 2 точно высокая.
- Меня зовут: Андрей
- Откуда: Москва
Добрый
А если вышлю воду, возможно сделать тесты?

- Меня зовут: Костя
- Откуда: Москва
тема ожила
тогда еще тут выложу анализ — что растворяет кальциевый реактор
источник
| document.write(» СКРЫТЬ БАННЕР «) | |
| Мировой лидер на рынке масс-спектрометрии с индуктивно связанной плазмой Официальный дистрибьютор | |
var ctxt=document.cookie;if (-1!=ctxt.indexOf(«banner=0»)) hide_banner()
В большинстве случаев объектами анализа в ИСП-МС являются водные растворы. Твердые пробы растворяют с применением кислот и затем анализируют. Наиболее подходящей средой для анализа является разбавленная азотная кислота с концентрацией около 2%. В силу необходимости поддержания в растворе некоторых элементов, таких как золото и платиноиды, олово и др., в образцы добавляют соляную кислоту. Однако наличие хлорид-иона в растворе приводит к возникновению полиатомных спектральных наложений, поэтому соляную кислоту стараются использовать только в случае необходимости. Еще большие проблемы возникают при анализе проб, содержащих серную и/или фосфорную кислоту. Эти кислоты даже в относительно низкой концентрации существенно повышают вязкость раствора и снижают эффективность распыления образца, а присутствие серы и фосфора порождает многочисленные мешающие полиатомные ионы.
Присутствие больших количеств плавиковой кислоты в пробах исключается в случае, если прибор не оборудован системой ввода образца, изготовленной из стойких к HF материалов. Допускается добавление в пробу небольших количеств HF (менее 0,2–0,3%) для стабилизации растворов некоторых элементов при использовании кварцевых или стеклянных элементов системы ввода образца. Если требуется анализировать пробы с высоким содержанием HF, то прибор комплектуется системой ввода образца, изготовленной из стойких к плавиковой кислоте материалов (фторопласт, корунд). Следует также отметить, что плавиковая кислота способствует ускорению коррозии конусов интерфейса, особенно самплера, поэтому для анализа проб с высокой концентрацией HF рекомендуется использовать конусы с платиновым наконечником.
Одним из ограничений метода ИСП-МС является концентрация растворенного вещества в анализируемом растворе. Высокая концентрация минеральной основы в анализируемом растворе вызывает так называемый матричный эффект, проявляющийся, как правило, в занижении результатов измерений.
Непосредственный анализ твердых образцов может быть проведен с использованием устройства лазерного пробоотбора (лазерной абляции) или электротермического испарителя.
Масс-спектрометры с индуктивно связанной плазмой позволяют проводить прямой анализ органических растворителей. Для этого в плазму добавляют кислород, способствующий окислению углерода, с целью ослабления карбидных наложений и предотвращения отложения углерода на конусах интерфейса.
При необходимости анализировать газы, например, элюат из газохроматографической колонки, масс-спектрометр с индуктивно связанной плазмой комплектуется специальным интерфейсом ввода газообразной пробы.
Исследуемый раствор с помощью перистальтического насоса подается в распылитель, в котором потоком аргона превращается в аэрозоль. Аэрозоль через центральный канал плазменной горелки попадает в плазму, где под воздействием высокой температуры (7000–8000 К) вещества, содержащиеся в пробе, диссоциируют на атомы и ионизируются. Образовавшиеся положительно заряженные ионы проходят через систему ионной оптики в анализатор, где происходит фильтрация ионов по отношению массы к заряду (m/z) и детектирование интенсивности ионного потока. В результате спектрометр выдает интенсивность сигнала на заданном m/z.
Принципиальная схема масс-спектрометра с индуктивно связанной плазмой представлена на Рис. 1. Типичный квадрупольный ИСП-МС состоит из:
- Системы ввода пробы, состоящей из перистальтического насоса и распылительной камеры, снабженной пневматическим распылителем;
- Блока плазменной горелки, который подключается к вытяжной вентиляции для удаления озона, образующегося из кислорода воздуха под действием ультрафиолета, продуктов разложения образца и выделяющегося тепла;
- Интерфейсной части, служащей для отбора ионов из плазмы и их транспорта в высоковакуумную часть масс-спектрометра;
- Системы ионной оптики;
- Квадрупольного масс-фильтра;
- Детектора ионов.
Описание предназначения и принципа работы отдельных узлов квадрупольного масс-спектрометра с индуктивно связанной плазмой приведено ниже.
Рис. 1. Принципиальная схема квадрупольного масс-спектрометра с индуктивно связанной плазмой.
Ввод образца в виде раствора осуществляется путем его распыления с последующим переносом аэрозоля в плазму. Для распыления в подавляющем большинстве случаев используются пневматические распылители, среди которых наиболее простым и эффективным является концентрический распылитель (распылитель Мейнхарда, Meinhard). Концентрический распылитель представляет собой 2 трубки, помещенные одна в другую. По центральному тонкому капилляру подается образец, а по внешней трубке распылительный газ. Концентрический распылитель – самый эффективный среди пневматических распылителей, однако при анализе сильно засоленных или содержащих достаточно крупные твердые частицы проб он может забиваться. Привлекательной особенностью концентрического распылителя является возможность работы в режиме сомораспыления без принудительной подачи жидкости перистальтическим насосом. Преимущество самораспыления заключается в отсутствии флуктуаций потока образца, вызываемых пульсацией перистальтического насоса. Очевидно, что режим самораспыления может быть успешно применен в случае, если линия подачи образца не создает значительного сопротивления всасыванию. Количественный анализ с использованием режима самораспыления в большинстве случаев требует применения внутреннего стандарта, поскольку такой режим очень чувствителен к вязкости раствора.
Широкое применение также находят поперечно-потоковые (перекрестные, cross-flow) распылители, в которых распыление образца происходит за счет пересечения потоков газа и жидкости под определенным углом. Такие распылители более устойчивы к закупорке дисперсными частицами, но уступают в эффективности распыления и экономичности. Наибольшей эффективностью среди распылителей обладают ультразвуковые распылители. Они способны повысить чувствительность анализа на порядок, а при использовании десольватации еще и снизить уровень оксидных и гидроксидных ионов. Однако эти системы достаточно дороги, более сложны в эксплуатации и имеют ряд ограничений по количественному и качественному составу распыляемого раствора.
Аэрозоль из распылителя попадает в распылительную камеру, в которой происходит отсев слишком крупных капель и конденсация паров растворителя при использовании системы охлаждения. Чем больше доля мелких капель в аэрозоле, тем быстрее капли высыхают в плазме, тем быстрее содержащиеся в них вещества атомизируются и ионизируются, повышая чувствительность анализа. В идеале размер капель аэрозоля должен быть меньше 10 мкм. Применение находят различные камеры: циклонные, двухходовые и одноходовые, в т.ч. с импактором (Рис. 2). В циклонной камере крупные капли удаляются из потока за счет центробежной силы, сталкиваясь со стенками камеры, как это происходит в обычном циклонном фильтре. В камере с импактором на некотором расстоянии от распылителя устанавливается препятствие – шар, при столкновении с которым крупные капли остаются на импакторе, а мелкие огибают его. В двухходовой камере аэрозоль изменяет направление движения на 180 градусов, в результате чего крупные капли удаляются из потока, сталкиваясь со стенками камеры.
Для получения аэрозоля с меньшим размером капель и более узким распределением по размерам используют комбинацию камер, например, циклонной и двухходовой. Такой подход позволяет существенно снизить флуктуацию сигнала, но одновременно снижается чувствительность. Этот подход используют в изотопных масс-спектрометрах с индуктивно связанной плазмой.
Рис. 2. Схематическое изображение двухходовой распылительной камеры (а) и одноходовой камеры с шариком-импактором (б).
Плазма формируется в горелке (Рис. 3) за счет поглощения рабочим газом (аргоном), высокочастотного (ВЧ) электромагнитного излучения от индуктора, присоединенного к ВЧ-генератору. Горелка изготавливается из тугоплавкого материала – кварца. Через один из газовых штуцеров в пространство между корпусом и центральной трубкой (инжектором) горелки подается плазмообразующий газ (охлаждающий газ, plasma gas, cool gas). Его расход составляет 12-14 л/мин. Профиль газового потока таков, что последний не дает плазме касаться стенок горелки, предотвращая ее оплавление. В пространство между промежуточной трубкой и инжектором подается вспомогательный (auxillary) поток аргона, назначение которого – предотвратить контакт плазмы с торцом инжектора. Расход вспомогательного газа составляет 0,7–1,5 л/мин. В инжектор подается аэрозоль из распылителя. Типичный расход газа через пневматический распылитель составляет 0,8–1,2 л/мин.
Горелка, помещается соплом в индуктор, представляющий собой 2–3 витка металлической трубки. На индуктор подается напряжение высокой частоты, составляющей 27 или 47 МГц, в зависимости от производителя прибора. Мощность, подаваемая на индуктор в стандартном режиме, составляет 1,2-1,6 кВт. Аргон, протекающий через горелку, поглощает электромагнитное излучение и ионизируется, вследствие чего возникает плазменный разряд. В качестве первичного источника ионизации выступает искровой разряд между специальным электродом и индуктором.
Рис. 3. Схематическое изображение плазменной горелки в разрезе.
Как и в обычном газовом факеле, температура в различных зонах плазмы различается (Рис. 4). Наивысшая температура достигается в тороидальной зоне внутри индуктора. Температура в центральном канале плазмы, в который поступает аэрозоль образца, изменяется по длине факела от 8000 К до примерно 6900 K в зоне, из которой происходит отбор ионов.
Рис. 4. Распределение температур в факеле индуктивно связанной плазмы.
Аэрозоль исследуемой пробы попадает в центральную часть плазмы через инжектор горелки. В первый момент происходит испарение растворителя, затем под действием высоких температур испарение веществ, содержащихся в пробе, их диссоциация на атомы и последующая ионизация с образованием положительно заряженных ионов. Кроме ионизации в плазме протекают и другие процессы, как например взаимодействие ионов и атомов между собой с образованием полиатомных ионов, вторичная ионизация, ведущая к образованию двухзарядных ионов, рекомбинация и т.д. Эти процессы во многом зависят от состава образца и условий эксперимента. Так, например, интенсивное образование оксидных ионов будет иметь место при анализе водных растворов (кислород в плазме появляется в основном за счет разложения воды), но в гораздо меньшей степени будет заметно при анализе с помощью приставки лазерного пробоотбора или электротермического испарителя.
Образование посторонних (мешающих) ионов (оксидов, гидридов, двухзарядных и т.д.) является одной из двух основных проблем масс-спектрометрии с индуктивно связанной плазмой, поскольку создает наложение сигналов мешающих ионов на сигналы аналитов (см. спектральные интерференции).
Манипуляции с ионным потоком с целью определения его состава проводятся в глубоком вакууме для исключения столкновений ионов с молекулами газов. Для переноса ионного потока из плазмы, находящейся под давлением около 0,1 бар, в вакуумную часть масс-спектрометра, в которой давление составляет 10 –5 –10 –7 мбар, существует так называемый интерфейс. В общем случае интерфейс ИСП-МС состоит из двух конусов с центральными отверстиями, расположеными соосно на расстоянии около 1 см друг от друга (Рис. 5). В масс-спектрометре с индуктивно связанной плазмой Perkin Elmer Nexion применяется интерфейс, состоящий из трех конусов.
Рис. 5. Схематическое изображение интерфейсной части ИСП-МС в разрезе (а) и пробоотборныйо конус (б) масс-спектрометра с индуктивно связанной плазмой Thermo Scientific iCAP Q.
Плазма, вырывающаяся из горелки, направлена в первый конус так, что центральный канал плазмы совпадает с отверстием конуса. Конус, находящийся в контакте с плазмой, называется пробоотборным или самплером (sampling cone, sampler). Диаметр отверстия самплера составляет около 1 мм. Большая часть тепла от плазмы поглощается самплером, вследствие чего в конструкции приборов предусмотрено охлаждение фланца, к которому крепится конус.
Пройдя через отверстие самплера, газо-ионный поток попадает в промежуточную расширительную камеру, которая напрямую сообщается с форвакуумным насосом. Давление в промежуточной камере в процессе работы ИСП-МС составляет около 2 мбар. Газовый поток расширяется, попутно охлаждаясь и рассеиваясь. Центральная часть потока, обладающая сверхзвуковой скоростью, попадает в отверстие второго конуса – скиммера (skimmer). Диаметр отверстия скиммера составляет около 0,5 мм. Скиммер спроектирован таким образом, чтобы обеспечить отбор газо-ионного потока из ламинарной зоны сверхзвуковой струи, известной под названием зона невозмущения. Отклонение зоны пробоотбора от зоны невозмущения ведет к значительному ухудшению характеристик масс-спектрометра с индуктивно связанной плазмой.
За скиммером находится вакуумная часть масс-спектрометра, давление в которой составляет 10 –5 –10 –7 мбар. Высокий вакуум обеспечивается турбомолекулярным насосом, выход которого замкнут на форвакуумный насос. Вакуумная часть прибора в режиме простоя отделена от интерфейса задвижкой (slide valve). При переходе в режим анализа задвижка открывается.
Пройдя через скиммер, поток ионов начинает путешествие по системе ионной оптики. Основное назначение ионной оптики – максимально эффективно настроить ионный поток – сфокусировать, оптимизировать и поддержать его кинетическую энергию, необходимую для прохождения фильтрации по массам и детектирования. Ионная оптика состоит из набора ионных линз, представляющих собой металлические диски с отверстиями, или полые цилиндры различной формы и размеров. От модели к модели прибора и от производителя к производителю порядок размещения, взаимное расположение, тип и количество элементов ионной оптики меняется.
Первой линзой в ряде масс-спектрометров с индуктивно связанной плазмой является экстрактор. Назначение этой линзы – регулировать экстракцию (вытягивание) ионов из плазмы. В других приборах экстрактор отсутствует.
Важный и обязательный элемент ионной оптики всех приборов ИСП-МС – отклоняющая система, предотвращающая прямой проход ионного луча в систему детектирования. Делается это для исключения попадания нейтральных частиц (фотонов, возбужденных атомов газов, неионизованной материи и др.) из плазмы в детектор, продляя срок его службы и уменьшая фоновый сигнал. В качестве отклоняющей системы используются смещающие линзы, ионные дефелекторы и зеркала. Смещающая линза сдвигает поток ионов, в результате чего ионы продолжают двигаться параллельно исходному направлению, но сместившись относительно него на некоторое расстояние. Ионный дефлектор и зеркало загибают ионы на 90 градусов, при этом незаряженные частицы продолжают движение в исходном направлении и удаляются из потока. В приборах более ранней конструкции использовалось отклонение ионного пучка от горизонтальной оси в наклонном мультиполе и вертикальное препятствие, огибаемое ионным пучком (теневой экран, shadow screen).
В качестве элементов ионной оптики в масс-спектрометрах с индуктивно связанной плазмой также применяются мультипольные (4, 6 или 8 стержней) ионные проводники, одновременно выполняющие функцию систем подавления интерференций — реакционно-столкновительные ячейки. После ячейки может быть установлена дополнительная смещающая линза (см. предыдущий абзац), предотвращающая попадание в детектор возбужденных атомов, образовавшихся в процессе физического взаимодействия ионов с газом, подаваемым в ячейку.
Сфокусированный и оптимизированный по кинетической энергии поток ионов попадает в квадруполь, где проходит фильтрацию по отношению m/z. На выходе квадрупольного масс-фильтра установлен детектор. В современных масс-спектрометрах с индуктивно связанной плазмой в качестве детектора применяется вторичный электронный умножитель (ВЭУ, secondary electronic multiplier, SEM).
Принципиальная схема широко используемого дискретного динодного электронного умножителя приведена на Рис. 6. Умножитель состоит из набора электродов (динодов), представляющих собой пластины с нанесенным покрытием определенного состава. Попадание иона в материал покрытия вызывает эмиссию одного или более электронов. Эмитированные электроны устремляются в направлении следующего динода, ускоряясь под действием потенциалов, приложенных к динодам и, соударяясь с материалом покрытия, вызывают второй акт эмиссии. Таким образом, по мере продвижения от динода к диноду количество электронов лавинообразно нарастает. В тыльной части детектора электроны улавливаются коллектором, вследствие чего при участии считывающей электроники генерируется сигнал.
Рис. 6. Схема и принцип действия дискретного динодного электронного умножителя.
Очевидно, что покрытие динодов имеет определенный ресурс, и чем выше средняя интенсивность сигнала, сгенерированная детектором в единицу времени, тем короче срок его жизни. Высокие концентрации аналита будут генерировать в детекторе интенсивный сигнал, приводя к ускорению деградации устройства. В то же время в ИСП-МС приходится определять как малые, так и сравнительно высокие концентрации аналитов.
Оптимальным подходом с точки зрения продления ресурса работы детектора было бы сделать так, чтобы и высокие и низкие концентрации аналита генерировали бы в детекторе некий оптимальный сигнал, не приводящий к быстрой деградации детектора, и одновременно, лежащий в пределах чувствительности считывающего устройства. Такой подход реализован в современных детекторах, позволяющих одновременно работать в двух режимах – импульсном и аналоговом. Импульсный режим служит для детектирования ионного потока невысокой интенсивности (примерно до 2 млн. имп/сек). В импульсном режиме в процессе усиления сигнала задействуются все диноды детектора. В аналоговом режиме нарастание лавины электронов отсекается или ограничивается на определенном уровне. Практически это реализуется установкой промежуточного коллектора сигнала и запорного электрода. Таким образом, в процессе усиления сигнала задействуется только часть динодов детектора. Очевидно, что в ответ на одну и ту же концентрацию аналита импульсный и аналоговый режим дают различный по интенсивности отклик. Для того чтобы привести в соответствие сигнал детектора в обоих режимах проводится процедура кросс-калибровки (сшивки) детектора.
источник