Водоподготовка и водный режим энергообъектов низкого и среднего давления. Справочник. Кострикин Ю.М. и др. 1990
| Водоподготовка и водный режим энергообъектов низкого и среднего давления. Справочник |
| Кострикин Ю.М., Мещерский Н.А., Коровина О.В. |
| Энергоатомиздат. Москва. 1990 |
| 252 страницы |
Описаны методы анализа исходных вод, приведены нормы качества обработанной воды, рассмотрены технологические процессы ее обработки. Рекомендованы способы оценки водного режима и его коррекции при эксплуатации. Изложены методы удаления отложений из оборудования, очистки и повторного использования сточных вод. Приведены данные по модернизации схем и оборудования водоподготовительных установок. Даны сведения о химических реагентах, способах их хранения, о водоподготовительном оборудовании и приборах. Для эксплуатационного и наладочного персонала промышленно-отопительных и теплоутилизационных котельных.
Раздел 1. Водоподготовка и коррекционная обработка воды
1.1. Общие сведения о воде
1.2. Анализы воды и их проверка
1.3. Роль примесей воды при ее использовании в энергетике
1.4. Предочистка. Методы и схемы обработки воды. Технологические и расчетные данные
1.5. Схемы обработки воды методами ионного обмена. Технологические и расчетные данные
1.6. Декарбонизация
1.7. Обработка воды методами осаждения
1.8. Обёзжелезивание артезианских вод
1.9. Обработка воды для тепловых сетей
1.10. Обработка конденсатов
1.11. Термический метод обессоливания
1.12. Деаэрация питательной и подпиточной воды
1.13. Показатели качества воды после отдельных стадий ее обработки
1.14. Выбор схем обработки воды
1.15. Расчет загрязнений стоков ВПУ при различных методах обработки воды
1.16. Коррекционные методы обработки воды
Раздел 2. Важнейшие сведения о пуске, наладке, эксплуатации водоподготовительных установок
2.1. Персонал химических подразделений энергообъектов
2.2. Обучение персонала и проверка его знаний
2.3. Технадзор за монтажом и приемка из монтажа
2.4. Составление исходной и оперативной технической документации
2.5. Составление инструкций
2.5.1. Перечень технологических инструкций по водоподготовке, водному режиму энергообъектов и контролю за ними
2.5.2. Должностные инструкции персонала
2.6. Порядок пуска оборудования, составление графика предпусковых и пусковых работ
2.7. Важнейшие требования, предъявляемые к оборудованию и сооружениям
2.8. Складское хозяйство и реагенты
2.9. Подготовка к пуску
2.10. Обработка охлаждающей воды
2.11. Борьба с коррозией
2.12. Водно-химические режимы паровых котлов и теплоутилизационных парогенераторов (ТУПГ)
2.13. Установление эксплуатационных норм водно-химического режима паровых котлов и ТУПГ
2.14. Краткие указания по внутреннему осмотру паровых котлов с целью оценки их водного режима
2.15. Удаление отложений с внутренней поверхности нагрева паровых и водогрейных котлов, теплообменных аппаратов и трубопроводов
2.16. Защита окружающей среды
2.17. Объем ревизий и ремонтов водоподготовительного оборудования
2.18. Общие указания по технике безопасности при работе на ВПУ
2.19. Условия эффективной эксплуатации ВПУ и ведения ВХР
Раздел 3. Организация и проведение физико-химического контроля за работой водоподготовительного оборудования и водно-химическим режимом работы энергообъектов
3.1. Значения и способы выражения результатов анализов воды, отложений, реагентов
3.2. Объем контроля за работой водоподготовительных установок и ведением водно-химического режима энергообъектов
3.3. Аналитические определения при анализе воды
3.4. Организация химических лабораторий
3.5. Отборы проб. Отборные, транспортирующие и охлаждающие устройства, организация отбора средних проб
3.6. Упрощенные методики определения качества воды энергообъектов. Визуально-органолептические определения. Количественные определения
3.7. Электрометрический контроль концентрации ионизированных примесей воды и растворов реагентов
3.8. Анализ сухих и жидких реагентов
3.9. Анализ товарных жидких и сухих технических реагентов
3.10. Отбор проб и упрощенный анализ отложений, образовавшихся на внутренних поверхностях теплоэнергетического оборудования
3.11. Упрощенные методы исследования свойств зернистых фильтрующих материалов
3.12. Применение индикаторов в тепловых сетях и конденсаторах для контроля процессов накипеобразования и коррозии
3.13. Вырезка контрольных участков и оценка состояния теплоэнергетического оборудования
3.14. Растворы аналитических реактивов
3.15. Выписка из сводных норм расхода этилового спирта на аналитическую работу на предприятиях Мосэнерго
3.16. Заменители лабораторных реактивов
Приложения:
Приложение 1. Некоторые химические соединения для обработки воды и химконтроля
Приложение 2. Свойства растворов, некоторых химических соединений (концентрация, нормальность, значения pH)
Приложение 3. Растворимость в воде составных частей воздуха
Приложение 4. Растворимость в воде реагентов для обработки воды
Приложение 5. Плотность растворов химических соединений
Приложение 6. Температура замерзания водных растворов
Приложение 7. Термодинамические свойства воды и водяного пара
Приложение 8. Качество воды природных источников
Приложение 9. Водно-химический режим паровых котлов (выписка из Правил Госгортехнадзора)
Приложение 10. Водно-химический режим котлов низкого давления, коммунальных установок и котлов-утилизаторов
Приложение 11. Водно-химический режим тепловых сетей
Приложение 12. Нормы качества перегретого пара (согласно ГОСТ 20995—75 и ПТЭ Минэнерго)
Приложение 13. Химические реагенты для обработки воды
Приложение 14. Конструктивные и технологические характеристики водоподготовительного и теплотехнического оборудования
Приложение 15. Защита оборудования от коррозии
Состояние подготовки воды на энергетических предприятиях определяет не только их экономичность, но и их экологическое совершенство, точнее, степень их экологической безопасности. Если современные крупные энергопредприятия обычно обеспечены достаточно квалифицированным персоналом, соответствующими инструкциями и технической документацией, то малые энергетические объекты, число которых чрезвычайно велико, совершенно лишены и того, и другого. Вследствие этого уровень эксплуатации водоподготовительных устройств на таких объектах крайне низок, значителен перерасход водообрабатывающих реагентов, вызывающий излишне высокое количество вредных в экологическом отношении сбросов, низка экономичность эксплуатации, необоснованно велик расход природных вод и т. д.
Настоящая книга представляет собой справочник, предназначенный в основном для эксплуатационного персонала отопительных паровых и водогрейных котельных и различных теплоутилизационных установок. Материал справочника может быть использован и эксплуатационным персоналом тепловых электрических станций ТЭС, ТЭЦ, КЭС и др. Однако главнейшими адресатами этого справочного материала являются мелкие котельные и отопительно-производственные объекты, разбросанные по территории СССР в промышленных и сельскохозяйственных населенных пунктах.
В справочном пособии изложены следующие вопросы.
В первом разделе даны сведения о воде, ее свойствах, требования, предъявляемые к ее качеству различными потребителями, методы и выбор схем обработки воды, технологические данные для расчета оборудования и показатели качества воды на различных стадиях ее обработки.
Второй раздел посвящен организации эксплуатации ВПУ, ведению водно-химических режимов энергообъектов.
В третьем разделе приведены сведения об организации химического контроля, оснащении химических лабораторий, краткие указания по выполнению простейших и важнейших определений качества воды, отложений, реагентов, даны критерии для оценки состояния теплотехнического оборудования с точки зрения коррозии, накипе- и солеотложений.
В приложениях даны сведения о природных водах и водных растворах, о реагентах, применяемых в водоподготовке, о нормах качества воды для котлов паровых и водогрейных, тепловых сетей, испарителей и др.
В справочнике даны также технологические и конструктивные характеристики водоподготовительного оборудования, дан перечень важнейшей литературы по водоподготовке и водно-химическому режиму.
Для выражения концентраций авторы справочного пособия пользовались не только молярными, но и эквивалентными величинами, а также производными от них, т. е. миллимольными, миллиэквивалентными, микроэквивалентными и т. д. Основанием для такого применения служило то обстоятельство, что молярные концентрации далеко не всегда применимы в технологии водоприготовления. Например, процессы ионирования, широко применяющиеся при обработке воды, сопровождаются обменом ионов с ионитными материалами, причем этот обмен происходит в эквивалентном соотношении. На один ион натрия поглощается не один ион кальция или магния а только половина, т. е. эквивалент.
С учетом всех этих факторов ряд организаций допустило применение эквивалентных единиц наряду с молекулярными.
Концентрации правильнее выражать применительно к массе, т. е. выражать в мг-экв/кг или в г/кг и т. д. Такое выражение концентраций единственно правильное, когда речь идет о примесях, содержащихся в насыщенном или перегретом паре. Однако в практике аналитической работы обычно не приходится взвешивать количества воды, отмеривая определенный объем ее, а концентрации относят к литру или к кубодециметру (мг/л или мг/дм3). Все эти узаконенные упрощения делают материал справочного пособия более доступным для тех лиц, кому оно предназначается.
Материал справочника распределяется между авторами следующим образом: разд. 1 составлен О.В. Коровиной, разд. 2 — Н.А. Мещерским, разд. 3 — Ю.М. Кострикиным и Н.А. Мещерским, приложения составлены Н.А. Мещерским. Авторы благодарят канд. техн. наук Г.П. Сутоцкого за огромную работу по рецензированию рукописи и ценные замечания, а также канд. техн. наук Н.П. Субботину за большую работу по редактированию книги.
источник
Материалы конференции «аналитический контроль качества воды в теплоэнергетике» москва кв ц «сокольники» конференц-зал №2
«Аналитический контроль качества воды в теплоэнергетике»
Москва, КВ Ц «Сокольники», конференц-зал №2
Опыт внедрения систем химико-технологического мониторинга (СХТМ) на ТЭС Российской федерации
Упрощенные методы оперативного химического контроля качества воды и пара.
Сравнительная оценка методов химического контроля качества воды.
В.Ф.Очков, Ю.А.Морыганова, В.Н.Кулешов
Обновленная версия книги Ю.М. Кострикина “Инструкция по анализу воды, пара и отложений в теплосиловом хозяйстве”
Б.Н.Дрикер, С.В.Смирнов, И.П.Сикорский
Аналитический контроль воднохимического режима на предприятиях теплоэнергетического комплекса
Б.Н.Шукайло, А.В.Талалай, В.И.Заволокин, М.В.Ивонин, К.Ю.Кудюков
Методика определения стабильности воды к выпадению карбоната кальция.
Проблемы материально-технического обеспечения деятельности аналитических лабораторий
Н.А.Белоконова, Л.В.Корюкова, И.О.Петухова
Возможности и перспективы использования методики оценки свойств органических соединений в водных растворах для решения производственных проблем энергетики
Л.В.Корюкова, Н.А.Белоконова, Л.С.Соловьев
Общие экологические проблемы энергетики и общества. Приборы для технологического и экологического контроля качества воды по содержанию органических веществ
О целесообразности контроля состояния ионитов при их применении в водоподготовке
Физико-химический анализ вод, используемых в системах энергоснабжения. Потенциометрические сенсоры для определения ионов кальция, жесткости и фосфатов (в том числе микроколичеств). Тест-методы полуколичественного определения ионов и активного хлора.
Опыт внедрения систем химико-технологического мониторинга (СХТМ) на ТЭС Российской федерации
Сметанин Денис Станиславович, МЭИ (ТУ), НПЦ «Элемент»
ООО «Научно-Производственный Центр «Элемент»
111250, Москва Е-250, Красноказарменная ул. 14
тел (095) 362-73-62 Тел/fax (095) 362-73-12
ООО “НПЦ “Элемент” основан в 1989г. сотрудниками Московского энергетического института (технического университета), имеющими 30-ти летний опыт работ в тепловой и атомной энергетике в области химической технологии внедрения систем химконтроля и химико-технологического мониторинга.
ООО “НПЦ “Элемент” совместно с МЭИ (ТУ) предлагают следующие работы:
Внедрение систем химико-технологического мониторинга с применением современных средств вычислительной техники
Проектирование систем химико-технологического мониторинга
Внедрение автоматизированных систем контроля и управления ВХР с применением современных средств вычислительной техники.
Входной контроль, поставка, наладка и сервисное обслуживание современных средств химконтроля
Создание и внедрение систем коррозионного мониторинга высокотемпературного и низкотемпературного тракта электростанций
Создание систем автоматического регулирования (САР) ввода корректирующих реагентов
Проектирование устройств подготовки пробы (УПП)
Изготовление, наладка и сдача в эксплуатацию диагностических комплексов мониторинга (с использованием амперометрических, кондуктометрических и потенциометрических безреагентных методов)
Разработка и внедрение пакета прикладных программ диагностики, анализа и прогнозирования водно-химического режима ТЭС
Разработка и внедрение пакета прикладных программ косвенных определений показателей качества ВХР
Создание тренажеров, моделирующих возможные нарушения ВХР
Внедрение систем экологического мониторинга с применением вычислительной техники водных объектов окружающей среды, включая стоки предприятий
Предпусковые и эксплуатационные химические очистки оборудования, консервация оборудования
Оптимизация водно-химического режима ТЭС и промышленных котельных с использованием СХТМ
уменьшение количества нарушений ВХР
снижение повреждаемости оборудования и аварийных остановов по вине ВХР
уменьшение расхода корректирующих реагентов
снижение расхода условного топлива
снижение недовыработки электроэнергии
уменьшение количества отложений на поверхностях нагрева
рост производительности труда персонала химических лабораторий
Цели и задачи создания систем мониторинга и управления химико-технологическими процессами на ТЭС
представление оперативной информации о состояния ВХР
минимизация нарушений ВХР
оптимизация химико-технологических процессов
снижение повреждаемости оборудования, связанной с нарушениями ВХР
снижение недовыработки электрической и тепловой энергии, связанной с нарушениями ВХР
Принципы создания систем мониторинга и управления химико-технологическими процессами на ТЭ
установка наиболее надежных и простых приборов автоматического химического контроля в наиболее уязвимых местах паро-водяного тракта;
максимально возможное использование имеющегося на энергетическом объекте парка приборов и помещений АХК;
использование теплотехнических параметров в СХТМ, влияющих на качество ВХР;
обязательное использование и ввод в ПЭВМ данных диагностического сменного и дневного лабораторного контроля;
совершенствование ВХР с использованием систем химико-технологического мониторинга и управления
создание и использование тренажеров, предназначенных для оперативного персонала и моделирующих возможные нарушения ВХР, их причины и методы устранения
Этапы построения систем мониторинга и управления химико-технологическими процессами на ТЭС
обследование водно-химического режима (ВХР) и формирование банка данных (БД) по существующему парку приборов для разработки технических предложений по внедрению СХТМ, включая требования к комплексу технических средств (КТС) и задач, решаемых системой
поставка, монтаж и наладка минимально необходимого КТС для «пилотной» СХТМ, включая приборы АХК, УСО и ПЭВМ; а также подключение существующих приборов АХК, технические характеристики которых позволяют использовать их в СХТМ
поставка и освоение персоналом пакета прикладных программ анализа ВХР с максимальным использованием данных ручных анализов и имеющегося парка приборов АХК
опытная эксплуатация «пилотной» СХТМ и внесение соответствующих изменений в задачи, решаемые СХТМ
разработка предложений на расширение СХТМ; поставка и наладка необходимого оборудования
адаптация и сдача в эксплуатацию полномасштабной системы
разработка комплекса задач для диагностики и прогнозирования состояния ВХР в темпе с процессом, включая советы оператору
В настоящее время СХТМ базируется на отечественных приборах автоматического и лабораторного химконтроля (фирмы “Техноприбор” г.Москва, “Взор” г. Н.-Новгород, “Инекотех” г. С-Петербург), котроллерах сбора данных.(фирма “ТЕКОН” г. Москва), УПП («СПЛАВ», г. Новгород).
Также ООО «НПЦ «Элемент» имеет опыт использования в СХТМ технических средств других производителей как ближнего, так и дальнего зарубежья.
В настоящее время ООО «НПЦ «Элемент» совместно с МЭИ проводит работы по внедрению СХТМ на Алексинской ТЭЦ, Воркутинской ТЭЦ-2, Казанской ТЭЦ-1, Набережно-Челнинской ТЭЦ, Петрозаводской ТЭЦ, Приморской ГРЭС, Северодвинской ТЭЦ-2, Сосногорской ТЭЦ, Ставропольской ГРЭС; Черепетской ГРЭС, Череповецкой ГРЭС.
Упрощенные методы оперативного химического контроля качества воды и пара.
115280, Москва, ул. Автозаводская, д.14/23
Упрощенные методы позволяют применять для ряда необходимых показателей качества оперативную или индикаторную систему контроля, которая дает возможность быстро получить ориентировочные данные, достаточные для немедленного вмешательства в состояние водно-химического режима с целью его корректировки.
Сравнительная оценка методов химического контроля качества воды.
Ковалева Н.Е., к.х.н., зав. лабораторией, ФГУП ИРЕА, Москва
107076, Москва, Богородский Вал, д.3
тел (095) 963-75-33 Тел/fax (095) 963-70-29
Известно, что при анализе одинаковых образцов даже в одинаковых условиях результаты не всегда получаются идентичными. Это имеет место из-за погрешностей, которые присущи каждой методике определения.
Помимо отклонений, вызываемых структурой анализируемого образца, влияние на получаемый результат оказывают оператор, применяемое оборудование, калибровка приборов, окружающие условия (температура, влажность и др.).
Важную роль в получении надежных результатов анализа играет правильный выбор метода анализа для данного конкретного случая. В стандартах, как правило, даются конкретные рекомендации по выбору метода в зависимости от цели исследования, тем не менее, в некоторых случаях аналитической службе предоставляется право самостоятельного выбора.
В настоящее время достаточно широко проводится техническое перевооружение аналитических лабораторий. В первую очередь это относится к внедрению средств вычислительной техники, применению инструментальных методов анализа методов. Однако роль химических методов в аналитическом контроле по-прежнему велика, ведь при выполнении анализов традиционными химическими методами не требуется дорогостоящая аппаратура. Более того, из-за меньшей зависимости результатов химических анализов от состава проб их широко используют для контроля качества анализов, выполняемых по инструментальной методике.
Однако, и химические методы анализа различны по своим возможностям. Основные ограничения применимости методов – сложность подготовки пробы, возможность устранения мешающих влияний других компонентов анализируемой смеси, чувствительность (минимальные концентрации определяемого вещества, при которых оказывается возможным его обнаружение) и точность, обусловленная чаще всего выбором метода контроля (визуальный, приборный и т.п.)
Обновленная версия книги Ю.М. Кострикина
“ Инструкция по анализу воды, пара и отложений в теплосиловом хозяйстве”
Очков В.Ф., к.т.н., доц., Морыганова Ю.А., к.х.н., доц., Кулешов В.Н., МЭИ (ТУ)
тел. 362-7171, e-mail: ochkov @ twt . mpei . ac . ru ; v.kul@.
Кафедра ТВТ МЭИ, совместно с ОАО “ВТИ”, ОАО “Фирмой ОРГРЭС” и ОАО “Мосэнерго” в 2003 году приступили к созданию обновленной версии книги Ю.М. Кострикина “Инструкция по анализу воды, пара и отложений в теплосиловом хозяйстве” .
Несмотря на солидный возраст, эта книга, по-прежнему остается основным источником информации для химиков-аналитиков на различных электростанциях. Описанные в ней методики являются основой для действующих на данный момент руководящих документов, используемых в химлабораториях электростанций и химических службах энергообъединений. Главная ценность книги в том, что она содержит множество теоретических пояснений и практических советов. Это значительно облегчает труд химиков-аналитиков, позволяет им правильно проводить анализы и получать достоверные результаты, особенно при возникновении внештатных ситуаций и всевозможных отклонениях от методики.
С момента последнего издания книги прошло достаточно много времени, некоторые методики анализа потеряли свою актуальность, а многие перспективные методы анализа, особенно инструментальные, в книге не описаны.
В связи с этим возникла острая необходимость создания обновленной версии книги “Инструкция по анализу воды, пара и отложений в теплосиловом хозяйстве” .
Обновленная версия не является изданием, принципиально отличающимся от книги Ю.М. Кострикина. Это существенно переработанный вариант оригинала, содержащий информацию о более широком круге методов анализа, и описание новых методик и приборов химконтроля, появившихся с момента последнего издания книги. Обновленная версия не является документом, заменяющим РД или ОСТы, действующие на данный момент, она должна использоваться параллельно с ними, являясь учебником, дополнением и справочником.
Список тем, необходимых для освещения в обновленной версии
краткие основы химического анализа;
основные принципы работы в химической лаборатории;
объемные (титримерические) методы анализа;
анализ отложений в пароводяных трактах электростанций;
анализ энергетических топлив;
анализ энергетических масел;
метрологические основы анализа воды, пара и отложений в теплосиловом хозяйстве;
анализ реактивов, используемых в теплосиловом хозяйстве.
1. Основной вариант обновленной версии – это печатное издание, т.е. книга, разделенная на главы (тома), издаваемые раздельно.
Каждый том посвящен отдельной теме из списка, приведенного выше, и состоит из трех основных частей . В первой части кратко приведены теоретические основы описываемого метода, возможные отклонения от методики, их причины и способы получения достоверных результатов в этих условиях. Во второй части приводятся оригиналы действующих РД и ОСТов, основанных на описываемом методе. Третья часть дает информацию о различных приборах и фирмах-поставщиках оборудования химконтроля, прошедших сертификацию.
После того, как будет закончена работа над последним разделом, их можно будет выпустить одной книгой, с учетом всех пожеланий и замечаний.
2. Второй вариант , выпускаемый параллельно с печатным изданием – это электронная версия, носителем которой является CD-ROM диск. Электронная версия позволяет оперировать гораздо большими объемами информации, помимо этого, позволяет использовать анимацию, различные контролирующие программы и тренажеры для лучшего усвоения информации. Существует возможность поставлять печатное издание и расширенную электронную версию совместно одним пакетом.
3. Третий вариант , создаваемый параллельно с первым и вторым – это электронный сайт в интернете, предоставляющий помимо описанной выше информации еще и возможность живого общения. Это даст возможность персоналу химлабораторий электростанций и химических служб энергообъединений обмениваться различными проблемами и способами их решения.
Кафедрой ТВТ созданы обучающие мультимедийные курсы, входящие в “Электронную Энциклопедию Энергетики” и посвященные следующим разделам:
объемные (титримерические) методы анализа;
— метрологические основы анализа воды, пара и отложений в теплосиловом хозяйстве.
Данные обучающие курсы могут быть взяты за основу планируемого печатного издания после учета всех замечаний и внесения изменений.
1. Необходимо пересмотреть уже созданные обучающие курсы и внести соответствующие поправки;
2. Параллельно с первым пунктом, создать новые обучающие курсы по оставшимся темам;
3. В процессе пересмотра уже созданного, приступить к отбору информации, которая войдет в печатное издание;
4. После этапа подготовки следует издание первого тома книги “Объемные методы анализа” или, например “Краткие основы химического анализа”;
5. На последних этапах издания первой части начинается подготовка к изданию второй части – “Хроматография” и т.д.
Как показал опыт, работа по созданию одного мультимедийного обучающего курса занимает 3-4 месяца. Можно предположить, что на создание каждого отдельного тома книги будет затрачено такое же время.
Аналитический контроль воднохимического режима на предприятиях теплоэнергетического комплекса
Дрикер Б.Н., д.х.н., проф., Смирнов С.В., Сикорский И.П., УГЛТУ, Екатеринбург
тел: (3432)355-12-69, ф: (3432)355-40-58
Внедрение современных технологий подготовки воды в теплоэнергетике, в частности, ингибиторов солеотложений, коррозии и биообрастаний на основе органофосфонатов, в значительной степени ужесточает требования к аналитическому контролю. Это связано с рядом обстоятельств, основными из которых являются:
качество воды в системах тепло- и горячего водоснабжения определяется качеством подпиточной воды;
реагенты, используемые в водоподготовке, – органофосфонаты и композиции на их основе, – хотя и применяются в субстехиометрических количествах, но при этом имеют относительно высокую стоимость и, кроме того, являются реагентами «порогового действия».
С учетом этого, наряду с традиционными методами аналитического контроля: определением жесткости, щелочности, содержания используемого для обработки воды органофосфоната и т.д., необходимо применение методов, которые сочетают высокую надежность и экспрессность, что позволяет в короткие сроки на основании результатов анализов проводить корректировку водно-химических режимов.
Электрод в форме диска, вращающийся в жидкости, отличается важной особенностью: его поверхность является равнодоступной в диффузионном отношении. По этой причине использование дискового электрода для определения интенсивности образующихся отложений и эффективности реагентной обработки воды представляется целесообразным, т.к. изменение конфигурации оборудования, а, следовательно, гидродинамических условий его эксплуатации, не имеет значения.
При использовании вращающегося дискового электрода в качестве катода, осаждение на нем карбоната кальция происходит в результате следующих реакций:
Таким образом, подщелачивание способствует образованию карбоната кальция на поверхности катода. Питание электрода осуществляется по гальваностатической схеме, что обеспечивает прохождение через раствор одного и того же количества электричества; при этом создаются условия постоянства электродвижущей силы процесса. Оптимальными условиями для изучения стабильности воды и эффективности ее реагентной обработки являются плотность тока 15—20 А/дм 2 и скорость вращения дискового электрода 1400 об/мин. При этих условиях относительная ошибка измерений не превышает 7%, а продолжительность анализа составляет не более 60 минут.
Для измерения скорости коррозии используется прибор «Aquamate». Определение коррозионной активности среды, находящейся в контакте с поверхностью металла, основано на использовании техники измерения линейного сопротивления поляризации. Для относительно постоянных значений коэффициентов Тафеля плотность поляризационного тока пропорциональна плотности тока коррозии. В технических водных системах проводимость растворов высока, а сопротивление растворов низко по сравнению с сопротивлением поляризации, поэтому плотность тока является достаточно точной мерой сопротивления поляризации и, соответственно, скорости коррозии. Поскольку двойной электрический слой на границе металл-жидкость имеет собственную характерную емкость, требуется определенное время для достижения установившегося значения уровня и тока коррозии. Как правило, этот интервал не превышает 30 минут.
Методика определения стабильности воды к выпадению карбоната кальция.
Шукайло Б.Н., Талалай А.В., Заволокин В.И., Ивонин М.В., Кудюков К.Ю., ООО «НПФ «М І ОР», г.Северодонецк, Украина.
93400, Украина, Луганская обл., г.Северодонецк, ул. Гоголя,16
Тел.: +38-050-598-12-39, ф: +38-064-524-30-14
В промышленной теплоэнергетике приходиться сталкиваться с проблемой выпадения карбонатно-кальциевых отложений. Умение прогнозировать этот процесс часто обуславливает безаварийность эксплуатации как водооборотных систем, так и водогрейных устройств (котлов, бойлеров).
Большинство исследователей при изучении процессов выпадения накипи предпочитает определять «термостабильность» воды. Суть дела сводится к следующему .
Исследуемая вода подвергается термостатированию или кипячению. Затем определяются химические характеристики воды и сравниваются с аналогичными для исходной воды. Чем меньше изменение (уменьшение) кальциевой жесткости и гидрокарбонатной щелочности, тем «стабильней» вода. Во время опыта также можно в исследуемую пробу воды помещать контрольную стеклянную пластину и затем исследовать количество и форму выпавших на ней кристаллов. Споры идут вокруг того следует ли кипятить воду или только нагревать, до какой температуры нагревать и сколько выдерживать , допускать ли свободную десорбцию углекислоты или препятствовать этому, фильтровать или нет исследуемую воду, учитывать или нет конечную величину рН воды и т.д.
Авторы длительное время занимались совершенствованием этого метода и достигли определенных успехов. Их основные усилия были направлены на повышение сходимости результатов. При определении «термостабильности» воды было предложено использовать нагрев воды в микроволновой печи. При этом гораздо легче соблюдать повторяемость главного условия эксперимента – равенство количества передаваемого пробе воды тепла от опыта к опыту. Кроме того самое серьезное внимание было уделено подготовке посуды для эксперимента: колбы ополаскивались соляной кислотой и отмывались дистиллированной водой, затем колбы ополаскивались исследуемой водой (с ингибиторами или без) для нейтрализации явления сорбции компонентов воды на стекле. В целом сходимость результатов оказалась на порядок лучше, чем при кипячении или термостатировании традиционным способом. Этим методом авторы пользуются около пяти лет.
Однако способ мало информативен и не позволяет получить количественную оценку «стабильности» воды.
Все это вынудило искать новые подходы при исследовании воды ( и ингибиторов накипеобразования). При разработке методики авторы отталкивались от мысли, что выпадение карбоната кальция из воды происходит при появлении в воде, содержащей кальций, карбонат-ионов:
Причем путь появления карбонат-ионов в воде значения не имеет. Так, при нагреве воды гидрокарбонаты превращаются в карбонаты:
2 НСО3 — = СО3 2- + Н 2 О + СО 2 (2)
Именно эта реакция происходит при нагреве воды в промышленных условиях и при определении «термостабильности». Однако, генерация карбонат-ионов может происходить и другим путем, например, при взаимодействии гидрокарбонатов с едкой щелочью:
НСО 3 — + ОН — = СО 3 2- + Н 2 О (3)
Это одна из реакций, происходящих в процессе реагентного умягчения воды. Исходя из этого вполне оправданной выглядела попытка изучения условий выпадения карбоната кальция не при кипячении (нагревании) воды, а при принудительном изменении ее кислотно-основного равновесия – подщелачивании. Как видно из уравнения (3) , повышение значения рН воды не происходит до тех пор, пока все гидрокабонатная кальцевая жесткость не превратиться в карбонат кальция – величина рН стабилизируется карбонатным буфером. И только после превращения гидрокарбонат-ионов в карбонат-ионы со связыванием их в СаСО 3 добавление щелочи приведет к возрастанию величины рН исследуемой воды. То есть согласно уравнениям (1) и (3) возрастание величины рН исследуемой воды при добавлении в нее щелочи будет тормозиться до полного расходования гидрокарбонатной жесткости. Если создать ситуацию, при которой реакция, описываемая уравнением (1) , будет ингибироваться, то при добавлении щелочи в исследуемой воде будут накапливаться карбонат-ионы взамен гидрокарбонат-ионов. То есть повышение величины рН среды будет происходить при добавлении щелочи сразу, а не после окончания реакций по уравнениям (3) и (1). Вышеописанную ситуацию можно создать при добавлении в исследуемую воду ингибиторов накипеобразования. Мерой «стабильности» воды, обработанной ингибиторами накипеобразования, таким образом, является количество едкой щелочи, необходимой для достижения заданного значения рН. Для обработанной ингибиторами воды это количество всегда будет меньшим.
Как видно из описания, для определения «стабильности» воды (или эффективности ингибиторов накипеобразования) необходим эксперимент с обработанной ингибиторами (исследуемой) водой и эксперимент с исходной водой (водой сравнения). Авторы попытались найти простой и понятный показатель, являющийся абсолютным, а не относительным критерием «стабильности» воды (или, говоря иначе, эффективности ингибиторов). Таким показателем может являться только поведение ионов кальция при различных условиях углекислотного равновесия. На этом моменте необходимо остановиться подробнее.
Произведение растворимости карбоната кальция сильно зависит от температуры, но при всех ее значениях невелико. Отношение произведения активностей ионов кальция и карбонат-ионов к ПРСаСО 3
и ли Са 2+ *[Са 2+ ] * C О3 2- [CO 3 2- ] (5)
где Са 2+ и C О3 2- — соответствующие коэффициенты активности и есть критерий «стабильности» воды. В литературе эта величина получила название «степень пересыщения». Это легкоопределяемый и понятный параметр.
Исходя из вышеизложенного, вкратце эксперимент выглядит следующим образом.
Аликвота исследуемой воды термостатируется при заданной температуре но не выше 40 0 С для предотвращение термического разложения гидрокарбонатов. Затем автоматической бюреткой дозируется раствор едкой щелочи до достижения заданной величины рН (авторы использовали значение 9,25 ед.рН). Регистрирующий прибор (планшетный самописец) рисует кривую, отображающую расход щелочи во времени на создание заданного значения рН в исследуемой воде и поддержании этого значения в течение 30 минут. В качестве основного оборудования авторы использовали титриметрическую установку Т-108 производства завода «ЗИП» г. Гомель, можно также использовать блок автоматического титрования БАТ-15 со стеклянной бюреткой и рН-метром. Разница между расходом щелочи для необработанной (исходной) воды и исследуемой (с ингибиторами) и есть количественный критерий оценки «стабильности» воды. Время эксперимента, выбрано исходя из реального времени пребывания воды в технологическом аппарате (бойлере, теплообменнике и т.д.) не превышающем нескольких минут. Выше описан эксперимент в «минимальном» виде, тем не менее, вполне достаточном для практических целей. В таком виде эксперимент требует поддержания постоянных температурных условий и величины рН. При расчете степени пересыщения эксперимент проводится только с исследуемой водой. Определение концентрации кальция в воде трудностей не представляет, определение содержания карбонат-ионов несколько сложнее. Соотношение между формами углекислоты в воде зависит от значения рН воды и в меньшей степени от температуры. Количество карбонат-ионов можно рассчитать, зная карбонатную щелочность воды и величину рН и используя справочные данные по соотношению форм углекислоты в воде при различных значениях рН, однако удобнее и проще пользоваться номограммами, опубликованными в литературе [1].
Ниже приводится пример конкретного эксперимента, проведенного с оборотной водой малого «чистого» оборотного цикла охлаждения электропечей Никопольского ферросплавного завода. Оборотная вода имеет следующие показатели :
Оборотная вода обрабатывалась композиций на основе фосфоновых кислот. Эксперимент проводился при температуре 40 0 С и величине рН 9,25 ед.рН. В опытах с необработанной оборотной водой расход 0,108N щелочи составил 2,70 0,05 мл на 100 мл воды, в опытах с обработанной водой эта величина равнялась 1,60 0,05 мл. Разница
относится на счет реакции :
Са 2+ + НСО 3 — + ОН — = СаСО 3 + Н 2 О (7)
которая в воде, обработанной ингибиторами накипеобразования тормозится.
Интересно сравнить характеристики обработанной и необработанной воды (фильтрованной пробы) после добавления щелочи.
Таблица 1. – Характеристики оборотной воды после подщелачивания
источник
Пробу отложений первоначально грубо измельчают, квартованием сокращают до нескольких граммов и растирают в агатовой или яшмовой ступке. Котельные отложения измельчают тщательно до мелкодисперсного состояния («пудры»).
Если при осмотре и измельчении пробы обнаруживаются частички сварочного грата, металлической стружки или иные загрязнения, их удаляют вручную. При этом нельзя пользоваться магнитом, так как окислы железа, содержащиеся в отложениях, также извлекаются магнитом.
В результате прокаливания масса накипи может уменьшиться или увеличиться. Уменьшение массы происходит вследствие потери массовой доли влаги, сгорания органических веществ, разложения карбонатов и т.д. Увеличение массы вызывается окислением кислородом воздуха закиси железа и металлической меди до их окислов.
Точную массу навески (0,5 — 1 г) отложений помещают в прокаленный и взвешенный фарфоровый тигель и прокаливают в муфельной печи при 800 — 850 °С до постоянной массы.
Прокаливание в никелевом, серебряном или платиновом тиглях недопустимо: никелевый тигель при этом сильно окисляется и загрязняет массу навески окисью никеля, серебряный может расплавиться, а платиновый будет испорчен, если в анализируемом материале относительно много металлической меди.
Если при этой температуре отложения плавятся, то берут новую массу навески и прокаливают при температуре 600 °С.
Тигель с массой навески устанавливают в холодную муфельную печь, постепенно повышая температуру до указанной, после чего прокаливают в течение 2 — 3 ч. Тигель с прокаленной массой навески охлаждают в эксикаторе, взвешивают и прокаливают вторично в течение 1 ч. Если масса изменится более чем на 0,5 мг, пробу прокаливают дополнительно.
Потерю при прокаливании в (ПП) вычисляют по формуле, %
где В1 — масса тигля с навеской до прокаливания, г;
В2 — масса тигля с навеской после прокаливания, г;
В — масса навески накипи, г.
Если имеется привес, его вычисляют, подставляя в формулу (1) разность (В2 — В1).
В зависимости от состава отложений перевод отобранной пробы в растворимое состояние производится обработкой ее водой, соляной кислотой, царской водкой или сплавлением со щелочью.
Отложения, образующиеся в трубках пароперегревателей и проточной части турбин, в основном состоят из водорастворимых соединений натрия, а также содержат нерастворимые в воде силикаты и продукты коррозии.
Отложения, образующиеся при нагревании воды в трубках конденсаторов турбин и различных подогревателях, так называемые низкотемпературные накипи, обычно нерастворимы в воде, но разлагаются соляной кислотой или царской водкой.
Отложения, образующиеся на внутренних поверхностях нагрева котлов, значительная часть которых нерастворима в кислотах, для перевода в растворимое состояние подвергаются сплавлению со щелочью.
Для определения массовой доли водорастворимых веществ массу навески воздушно-сухих измельченных отложений (0,5 — 1 г) помещают в химический стакан, вливают в него 50 см 3 горячей дистиллированной воды, образовавшиеся комочки раздавливают и растирают стеклянной палочкой и кипятят жидкость на слабом огне в течение 10 — 15 мин, периодически помешивая ее.
Содержимое стакана количественно переводят в мерную колбу вместимостью 500 см 3 , смывая туда также оставшиеся частички отложений со стенок стакана струей горячей дистиллированной воды. Жидкости в закрытой колбе дают остыть, после чего доливают до метки дистиллированной водой, содержимое колбы периодически тщательно перемешивают и оставляют отстаиваться. Из хорошо осветлившейся жидкости, не перемешивая ее, отбирают пробы для определения щелочности, жесткости, кальция, магния, сульфатов, хлоридов, силикатов, фосфатов по методам, изложенным в [1 — 4], и натрия пламяфотометрически [1, 2] в соответствии с инструкцией, прилагаемой к прибору.
Если нет полного осветления жидкости, то анализы выполняют из отфильтрованной пробы.
Для анализа нерастворимой в воде части отложений используется осадок, полученный после отделения водорастворимых веществ фильтрованием через беззольный фильтр, прокаливанием и последующим растворением осадка в соляной кислоте, царской водке или сплавлением со щелочью.
Если анализируются отложения с высоким содержанием не растворимых в воде веществ, то отложения для перевода в растворимое состояние обрабатывают кислотой или подвергают предварительному сплавлению со щелочью.
Массу навески (0,5 — 1 г) воздушно-сухих отложений или полученных после определения массовой доли потери при прокаливании количественно переносят в химический стакан, смывая 30 — 50 см 3 разбавленной (1:1) соляной кислотой. Жидкость кипятят около 10 мин, не допуская бурного течения реакции во избежание разбрызгивания раствора. Если отложения полностью не растворятся в соляной кислоте, то добавляют 0,33 объема концентрированной азотной кислоты, создавая таким образом раствор царской водки. В случае неполного растворения отложений в царской водке производят сплавление со щелочью отфильтрованного нерастворенного остатка или сплавляют со щелочью всю массу навески отложений.
Разложение отложений для полного перевода их в растворимое состояние может быть выполнено сплавлением с едкими натром или калием, содой, или смесью соды с поташом в соотношении 1:1. Сплавление с едким натром более эффективно, оно выполняется при 500 — 600 °С в течение 30 — 40 мин; сплавление с содой нужно выполнять в платиновом тигле [1 — 3, 5] при температуре 900 — 950 °С в течение 2 — 3 ч. Поэтому предпочтительно производить разложение отложения сплавлением с едким натром.
Сплавление с едким натром выполняют в никелевых или серебряных тиглях. Применять для этого платиновые тигли нельзя, так как при сплавлении с едким натром происходит заметное растворение платины, что приводит к ее потере и затрудняет дальнейшее выполнение анализа.
Никелевый или серебряный тигель, если он еще не был в употреблении, на одну треть заполняют твердым едким натром и ставят на 30 — 40 мин в муфельную печь, нагретую до 400 — 600 °С. После этого тигель извлекают, дают остыть и выщелачивают получившийся сплав дистиллированной водой.
В чистый тигель берут массу навески сухого едкого натра (квалификации «х.ч.») из расчета 3 — 4 г на 1 г массы навески отложений, помещают в холодную муфельную печь и нагревают до 400 — 600 °С. Когда щелочь расплавится (обычно за 2 — 3 мин), тигель вынимают из муфельной печи. В остывший тигель на затвердевшую щелочь переносят массу навески прокаленной накипи из фарфорового тигля. Количество перенесенной накипи проверяют взвешиванием фарфорового тигля. Если требуется ускорить выполнение анализа, производят сплавление со щелочью воздушно-сухой пробы отложений, а потерю при прокаливании определяют из отдельной массы навески.
Тигель со щелочью и массой навески отложений помещают в муфельную печь, повышают температуру до 500 — 600 °С и выдерживают 30 — 40 мин. Затем тигель из печи осторожно вынимают. Если сплав однородный, то сплавление считают оконченным. В противном случае выдерживают тигель в муфеле еще 10 — 20 мин (обычно этого не требуется, если накипь была хорошо измельчена).
Извлечение сплава производят следующим путем. Соляная кислота растворяет металлический никель. Концентрированный раствор щелочи, особенно горячий, воздействует на фарфор и стекло. Поэтому необходимо избегать воздействия кислоты на никель и, по возможности, сокращать продолжительность действия щелочи на фарфор и стекло. Учитывая это, извлечение сплава из никелевого тигля рекомендуется производить водой. В еще теплый, но не горячий тигель с массой навески наливают дистиллированную воду примерно до половины тигля и осторожно подогревают, помешивая содержимое стеклянной палочкой. Жидкость из тигля выливают по папочке в фарфоровую чашку, в которую предварительно наливают 20 — 30 см 3 соляной кислоты, разбавленной 1:1. Если сплав полностью не растворился, эту операцию повторяют до полного растворения и перенесения сплава в фарфоровую чашку. Стеклянную палочку не следует оставлять в тигле, а надо, помешав его содержимое, вынимать ее и класть в фарфоровую чашку, куда переносится растворяющийся сплав. Сливать жидкость из тигля в чашку надо быстро, не допуская выливания жидкости на наружную поверхность тигля. Если это произошло, надо смыть эти капли небольшим количеством дистиллированной воды в фарфоровую чашку. Перед началом выщелачивания сплава наружную поверхность тигля нужно промыть дистиллированной водой. При сплавлении пробы отложений с едким натром и вымывании сплава водой (без добавления кислоты) из никелевого тигля переходит в раствор незначительное количество никеля, не мешающее выполнению нижеприведенных анализов.
Соляная кислота почти не действует на серебро. Серебряный тигель, обмыв его наружную поверхность, можно опускать в разбавленную соляную кислоту и кипятить в ней до полного растворения сплава.
Если разложение отложений производится водой или соляной кислотой, то при определении содержания меди и железа для их окисления в анализируемые пробы исходного раствора следует добавлять несколько кристаллов персульфата аммония, несколько капель пергидроля или азотной кислоты.
Раствор отложения, полученный обработкой массы навески соляной кислотой или царской водкой или после сплавления пробы со щелочью, выпаривают в фарфоровой чашке на кипящей водяной бане досуха. Затем в чашку наливают 10 — 15 см 3 концентрированной соляной кислоты и выпаривают до получения сухого остатка. Такую обработку соляной кислотой для перевода кремниевой кислоты в нерастворимое состояние выполняют 2 — 3 раза. После этого в охлажденную чашку вливают 5 — 10 см 3 концентрированной соляной кислоты и 15 — 20 см 3 горячей дистиллированной воды, нагревают на водяной бане, перемешивают, добиваясь полного растворения солей, и через несколько минут фильтруют через плотный беззольный фильтр «синяя лента». Для ускорения фильтрования на фильтр помещают небольшое количество фильтробумажной (мацерированной) массы [6]. Фильтрат собирают в мерную колбу вместимостью 500 см 3 . Фильтрат должен быть совершенно прозрачным. Стенки чашки и осадок на фильтре промывают кипящей дистиллированной водой, подкисленной соляной кислотой (2 — 3 см 3 концентрированной соляной кислоты на 250 см 3 ), до отрицательной реакции на трехвалентный ион железа (проба с 10 %-ным раствором роданистого аммония) и затем горячей дистиллированной водой до отрицательной реакции на ион хлора (проба с 1 %-ным раствором нитрата серебра, подкисленным азотной кислотой). Чашку вытирают кусочками беззольного фильтра, которые переносят на фильтр. Промытый осадок вместе с фильтром слегка подсушивают в воронке в сушильном шкафу и переносят в прокаленный и взвешенный фарфоровый тигель, доведенный до постоянной массы, высушивают, озоляют и прокаливают в течение 1,5 — 2 ч при температуре 950 — 1000 °С с повторным прокаливанием в течение 30 — 40 мин до постоянной массы.
Содержание кремнекислоты в процентах в отложении вычисляют по формуле
где В2 — масса тигля с двуокисью кремния, г;
В1 — масса пустого тигля, г;
В — масса навески накипи, г.
Осадок кремнекислоты бывает загрязнен следами железа, алюминия и другими примесями. Если анализ необходимо выполнить точно, то определяют содержание кремнекислоты по потере массы, удалив кремний в виде летучего тетрафторида SiF 4 , что выполняют только в платиновом тигле. Для этого после взвешивания прокаленный осадок двуокиси кремния смачивают в тигле несколькими каплями дистиллированной воды, вводят около 0,5 см 3 концентрированной серной кислоты и, когда тигель остынет, добавляют 5 — 7 см 3 чистой концентрированной плавиковой кислоты и содержимое тигля осторожно перемешивают вращением тигля. Раствор в тигле медленно выпаривают сначала на водяной бане, потом на плитке. После прекращения выделения белого дыма (серного ангидрида SO 3 ) тигель с остатком прокаливают при 950 — 1000 °С в течение 30 — 40 мин, охлаждают в эксикаторе и взвешивают. Когда имеются опасения, что не вся двуокись кремния превратилась в тетрафторид, обработку кислотами с выпариванием и прокаливанием повторяют. Остаток в тигле, если масса его значительна, растворяют в соляной кислоте, и раствор присоединяют к фильтрату от кремнекислоты.
Содержание кремнекислоты ( SiO 2 ) вычисляют по формуле, %
где B 1 — масса тигля с осадком загрязненной двуокиси кремния, г;
В2 — масса тигля после удаления тетрафторида SiF 4 , г;
В — масса навески накипи, г.
Остаток после удаления кремнекислоты обычно небольшой. Поэтому проверочное удаление кремнекислоты производят редко.
Фильтрат от кремнекислоты, содержащий все катионы, а также анионы фосфорной и серной кислот, которые были в накипи, доводят дистиллированной водой в мерной колбе вместимостью 500 см 3 до метки и перемешивают. Полученный раствор именуется исходным или основным раствором накипи. Его используют для определения других компонентов накипи.
Содержание железа определяется титрованием трилоном Б в присутствии сульфосалицилат-ионов при значении рН раствора 1-2. Присутствие ионов кальция, магния, алюминия, цинка, марганца, никеля, фосфатов, сульфатов не мешает выполнению анализа. Медь не образует с сульфосалицилат-ионами окрашенного соединения, но в присутствии железа при значении рН — 2 почти количественно титруется трилоном. При значении рН = 1 медь значительно меньше завышает результаты определения содержания железа. При значении рН менее 1 разрушение трилоном соединений железа с индикатором идет медленно, и переход окраски индикатора затягивается. При высокой концентрации сильных кислот подавляется диссоциация слабой сульфосалициловой кислоты, и окрашенное в вишневый цвет комплексное соединение трехвалентного железа не образуется.
Влияние меди на определение массовой доли железа существенно уменьшается, если анализируется раствор со значением рН = 1 и с небольшим количеством индикатора сульфосалициловой кислоты.
Так, например, при анализе раствора с рН = 1, введении 0,1 см 3 20 %-ной сульфосалициловой кислоты и массовой доле меди в пробе 3 — 10 мг завышаются результаты определения железа соответственно примерно на 0,2 — 0,6 мг, что при массе навески отложения 1 г, массовой доле меди 5 — 50 %, объеме исходного раствора 500 см 3 и объеме пробы на анализ 10 см 3 завышаются данные определения массовой доли железа в отложениях на 1 — 3 %.
Приведенные данные указывают, что обычно можно определять массовую долю железа, не отделяя его от меди. Если же массовая доля меди относительно велика и анализ требуется выполнить по возможности более точно, то определение железа производится после осаждения его аммиаком, отделения фильтрованием и растворения соляной кислотой. Обычно для полного отделения железа от меди производится двукратное осаждение и отделение полуторных окислов добавлением аммиака в небольшом избытке [1-3]. При этом осадок полуторных окислов может быть использован для трилонометрического определения массовой доли железа и затем алюминия [1], а фильтрат — для определения суммарной массовой доли меди и цинка, а затем суммарной массовой доли кальция и магния [3].
Отделение железа от меди аммиаком можно значительно ускорить однократным осаждением железа, если добавлять аммиак в большом избытке и операции осаждения и фильтрования объединить в одну [7] как описано ниже.
Определение массовой доли железа после его отделения от меди лучше производить при значении рН ≈ 2, так как при этом переход окраски индикатора более четкий, чем при рН = 1.
Ввиду малой скорости взаимодействия железа с трилоном Б раствор нагревают перед титрованием до 60 — 70 °С.
Трилон Б, 0,05-молярный (0,1 — нормальный) или 0,005 — молярный (0,01 — нормальный) растворы.
Сульфосалициловая кислота, 20 %-ный раствор.
Соляная кислота, раствор 1:1.
из исходного раствора отложений отбирают в коническую колбу в зависимости от предполагаемой массовой доли железа 10 — 25 см 3 и добавляют для гарантии полного окисления железа несколько капель концентрированной азотной кислоты, перекиси водорода или несколько кристаллов персульфата аммония. Определить по индикаторной бумаге с достаточной точностью значение рН раствора, равное 1, невозможно, поэтому доведение его до указанного значения выполняют следующим путем. Пробу нагревают до кипения и медленно при перемешивании добавляют разбавленный (1:1) раствор аммиака до начала образования осадка гидроксида железа. Добавляют 10 см 3 раствора 1 — нормальной соляной кислоты и, когда осадок гидроксида железа растворится, дистиллированной водой доводят объем до 100 см 3 . После этого анализируемый раствор нагревают до 60 — 70 °С, добавляют 0,1 см 3 20 %-ного раствора сульфосалициловой кислоты и титруют 0,05-молярным или 0,005-молярным раствором трилона Б до изменения вишневой или розовой окраски в желто-зеленую или до обесцвечивания (трилонат железа имеет слабую желто-зеленую окраску, заметную лишь при высокой концентрации железа).
Если оттитрованный раствор при стоянии медленно опять розовеет, что может быть из-за наличия меди, на это не следует обращать внимания.
При значительной массовой доле меди производят осаждение железа аммиаком и отделяют его фильтрованием. Для фильтрования применяют обычную воронку с беззольным фильтром «красная лента» или «белая лента» или лучше воронку цилиндрической формы с крупнопористой пластинкой, на которую помещают слой ваты высотой 5 — 10 мм. Нижнее отверстие воронки закрывают полиэтиленовой или резиновой трубкой с винтовым зажимом и устанавливают в штатив над стаканом. В воронку наливают 5 — 10 см 3 концентрированного аммиака и в зависимости от предполагаемой массовой доли железа 10 — 25 см 3 исходного раствора отложений и оставляют на 10 мин. Затем открывают затвор, отфильтровывают осадок и промывают 3-4 раза горячим раствором аммиака 1:20. Воронку с промытым осадком помещают над конической колбой, осадок растворяют около 5 см 3 горячей соляной кислоты 1:1 и промывают несколько раз горячей дистиллированной водой. Доводят объем пробы дистиллированной водой до 100 см 3 и добавляют раствор аммиака 1:1 до значения рН ≈ 2. После этого производят определение содержания железа, как указано выше.
Массовую долю железа в пересчете на оксид железа ( Fe 2 O 3 ) вычисляют по формуле, %
где а — расход раствора трилона Б на титрование, см 3 ;
М — молярность раствора трилона Б;
К — поправочный коэффициент к данной молярности;
V исх — общий объем исходного раствора отложений, см 3 ;
V пр — объем исходного раствора отложений, взятый для анализа, см 3 ;
В — масса навески отложений, г;
79 ,85 — эквивалентная масса Fe 2 O 3 в реакции с трилоном Б, равная половине молекулярной массы.
Определение массовой доли железа из исходного раствора накипи можно выполнить также с помощью автоматического титратора (Т-107 или Т-108). Автоматические приборы для титрования позволяют повысить точность и объективность получаемых результатов, а также сократить время анализа.
Метод основан на том, что в среде двухзамещенного аммония лимоннокислого (рН ≈ 5) в присутствии индикатора 1-(2-пиридилазо) — 2-нафтол (ПАН) трилоном Б количественно титруется сумма ионов Cu 2+ и Zn 2+ [ 6 , 9]. В отличие от меди цинк не образует с ПАН в указанной среде окрашенного соединения, поэтому анализ выполним лишь в присутствии меди (11). Экспериментально установлено, что если массовая доля меди в пробе менее 0,1 мг, могут получаться заниженные результаты определения суммарной массовой доли меди и цинка. Во избежание этого, если неизвестно, что меди достаточно, вводят раствор трилоната меди (или точное количество меди, которое затем учитывают, вычитая из расхода трилона на титрование расход его на введенную медь).
Анализ выполним в присутствии железа, алюминия, кальция, магния, никеля. Железо связывается в нитратный комплекс и три лоном Б не титруется. При массовой доле железа в титруемом растворе более 10 мг переход окраски отмечается менее четко, так как в указанной среде наличие железа придает раствору желтую окраску. Ионы Mn 2+ замедляют переход окраски индикатора. В их присутствии во избежание получения завышенных результатов анализа титрование раствором трилона Б нужно производить медленно, особенно вблизи эквивалентной точки. Присутствие кальция, магния, алюминия не мешает выполнению анализа. Присутствие никеля не мешает выполнению анализа, если учитывать следующее. При комнатной температуре в среде двухзамещенного лимоннокислого аммония взаимодействие трилона Б с никелем происходит настолько медленно, что в его присутствии наблюдается четкий переход окраски индикатора при окончании титрования цинка и меди. Окраска оттитрованной пробы, содержащей никель, постепенно изменяется, на что не следует обращать внимания. С повышением температуры реакция взаимодействия трилона Б с ионами никеля ускоряется, поэтому для определения массовой доли суммы ионов Cu 2+ и Zn 2+ в присутствии никеля температура титруемого раствора не должна превышать 20 — 25 °С.
Метод позволяет определить суммарную массовую долю меди и цинка 1 мг в пробе с относительной ошибкой около 10 %. При наличии в отложениях марганца ошибка анализа увеличивается.
Трилон Б, 0,005-молярный или 0,05-молярный раствор.
Аммоний лимоннокислый двухзамещенный, 20 %-ный раствор
Смешанный индикатор ПАН — смесь 0,08 г ПАН в 80 см 3 этилового спирта и 0,02 г метиленового голубого (метиленового синего) в 20 см 3 спирта или 0,1 %-ный спиртовой раствор ПАН.
Раствор трилоната меди. Смешивают точно эквивалентные количества 0,1-нормадьных растворов сульфата меди и трилона Б.
отбирают в коническую колбу 10 — 20 см 3 исходного раствора отложения, разбавляют дистиллированной водой до 100 см 3 и добавлением аммиака доводят до рН ≈ 3. Значение рН определяют по индикаторной бумаге. Добавляют 10 см 3 раствора двухзамещенного аммония лимоннокислого, 1 — 2 см 3 трилоната меди, 5 — 7 капель индикатора и медленно при интенсивном перемешивании титруют раствором трилона Б до изменения окраски: из фиолетово-сиреневой в желто-зеленую при применении смешанного индикатора; когда применяют ПАН без метиленового голубого окраска изменяется из фиолетово-красной в желтую.
Медь и цинк имеют близкие по значению атомные массы, поэтому, если не определяют раздельно массовые доли меди и цинка, их общую массовую долю в пересчете на оксиды рассчитывают по формуле, %
где а — расход раствора трилона Б на титрование, см 3 ;
М — молярность раствора трилона Б;
K — поправочный коэффициент к данной молярности;
80 — молекулярная масса (усредненная) 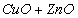 .
.
В среде бифторида аммония (рН ≈ 3,7) в присутствии индикатора ПАН и ионов меди (II) количественно титруются трилоном Б ионы меди и цинка, а затем после нагревания титруется никель [8].
Если меди относительно мало, то во избежание получения заниженных результатов анализа вводят трилонат меди (или точное количество меди, вычитая расход трилона при титровании на введенную медь).
Анализ выполним в присутствии железа, кальция, магния, алюминия, марганца.
Фтористые соли разъедают стекло, поэтому для анализов с их применением следует выделить специальную посуду. Однако ввиду отсутствия полиэтиленовых конических колб для титрования допускается использование стеклянных. С одними и тени же стеклянными колбами можно работать несколько месяцев.
Метод позволяет определить суммарную массовую долю меди и цинка 1 мг в пробе с относительной ошибкой около 5 %.
Трилон Б, 0,005-молярный раствор.
Бифторид аммония, 20 %-ный раствор (приготавливать и хранить в полиэтиленовой посуде).
Раствор индикатора ПАН (см. п. 6).
Раствор трилоната меди (см. п. 6).
отбирают в коническую колбу 10 — 20 см 3 исходного раствора отложения, разбавляют дистиллированной водой до 100 см 3 и добавлением аммиака доводят до рН ≈ 3 (определяют рН по индикаторной бумаге). Затем добавляют 10 см 3 раствора бифторида аммония (отмеряют небольшим протарированным сосудом из оргстекла или полиэтилена), 1 — 2 см 3 трилоната меди, 5 — 7 капель индикатора и титруют трилоном Б до перехода окраски: из фиолетово-сиреневой в желто-зеленую при применении смешанного индикатора ПАН; когда применяют раствор ПАН без метиленового голубого, окраска изменяется из красно-фиолетовой в желтую.
Расход трилона Б на титрование соответствует суммарной массовой доле меди и цинка. Если при стоянии раствора постепенно появляется розовый оттенок вследствие медленного взаимодействия индикатора с никелем, на это не следует обращать внимания.
Для определения массовой доли никеля раствор, полученный после определения суммарной массовой доли меди и цинка, нагревают до 70 — 80 °С и, если при этом раствор окрасится в фиолетово-сиреневый или красно-фиолетовый цвет, его титруют трилоном.
Суммарную массовую долю меди и цинка вычисляют по формуле (5).
Массовую долю никеля в пересчете на оксид никеля ( NiO ) вычисляют по формуле, %.
где δ — расход трилона Б на титрование подогретой пробы, см 3 ;
74 ,71 — молекулярная масса NiO .
Массовую долю меди определяют иодометрически, а затем в той же пробе комплексометрически определяют массовую долю цинка [9, 10].
Двухвалентная медь в слабокислой среде восстанавливается йодистым калием до одновалентной, выделяя из йодистого калия эквивалентное количество йода. Выделившийся йод титруют тиосульфатом натрия в присутствии крахмала, добавляемого в конце титрования.
Для того, чтобы реакция восстановления меди и выделения йода протекала количественно, йодистый калий вводят в большом избытке. Реакцию нужно проводить в слабокислой среде. С повышением рН раствора реакция восстановления меди замедляется и может не дойти до конца. В сильно кислой среде происходит окисление йодид-ионов кислородом воздуха. Требуемая кислотность создается добавлением бифторида аммония или бифторида калия к исходному раствору отложений, предварительно нейтрализованному до значения рН ≈ 3.
Йодометрическому определению массовой доли меди мешают окислители, в том числе трехвалентное железо, которое обычно присутствует в отложениях. Бифторид связывает железо во фторидный комплекс, не мешающий определению меди.
Наличие алюминия, кальция, магния, марганца, никеля, фосфатов в тех количествах и соотношениях, какие обычно бывают в исходных растворах котельных накипей, не мешает йодофторидному определению массовой доли меди.
Анализ выполняют из исходного раствора, получаемого сплавлением массы навески отложения с едким натром в никелевом или серебряном тигле.
Если сплавление массы навески отложения производится в платиновом тигле с карбонатами натрия и калия, то при введении йодистого калия для определения массовой доли меди наблюдается появление не желтой, а желто-вишневой окраски, более яркой при большей массовой доли меди. При титровании пробы тиосульфатом натрия в присутствии крахмала сперва исчезает синяя окраска, остается более слабая желто-вишневая, которая тоже обесцвечивается при дальнейшем добавлении тиосульфата натрия; в этом случае нет уверенности в правильности определения точки конца титрования.
Метод позволяет определить массовую долю меди в пробе с относительной ошибкой около 7 %.
Для определения массовой доли цинка после определения массовой доли меди рН раствора доводят добавлением аммиака до значения 4, проверяя его для большей точности не по индикаторной бумаге, а по так называемому вечернему индикатору. Реакция взаимодействия двухвалентной меди с йодистым калием обратима и при повышении значения рН сдвигается в сторону образования двухвалентной меди. При рН = 4 небольшая часть восстановленной меди опять переходит в двухвалентную, но наличие ее дает возможность определять массовую долю цинка титрованием трилоном Б с индикатором ПАН, так как в создаваемой среде и при указанном значении рН цинк не дает с ПАН красного окрашенного соединения.
Анализами растворов, не содержащих цинка, и растворов с известной массовой долей цинка установлено, что при первоначальной массовой доле в пробах двухвалентной меди в количестве 3 — 10 мг (когда на титрование йода, выделившегося при добавлении йодистого калия расходуется 0,5 — 1,5 см 3 0,1-нормального тиосульфата натрия) при добавлении избытка 0,5 см 3 раствора тиосульфата натрия и доведении рН раствора до значения 4 опять появляется двухвалентная медь в количестве 0,13 — 0,19 мг (на ее титрование расходуется 0,4 — 0,6 см 3 0,005-молярного раствора трилона Б). Если первоначальное количество меди меньше указанного и в пробу после доведения до рН ≈ 4 добавить 1 — 2 см 3 0,005-молярного раствора сернокислой меди, то в анализируемом растворе остается двухвалентная медь в том же количестве (0,13 — 0,19 мг).
Для определения массовой доли цинка и внесения правильной поправки на медь нужно по возможности точно доводить значение рН до 4, так как с повышением его значения больше меди переходит в двухвалентную, что завышает результаты анализа определения цинка, а при меньшем его значении будут получены заниженные результаты. Если переход окраски индикатора нечеткий и нет уверенности, что pH раствора приближается к 4, то массовую долю цинка можно вычислить по разности между суммой меди и цинка формулы (5 и 6) и меди формула (7).
При массовой доле в пробе кальция более 5 мг, магния более 3 мг получаются заниженные результаты определения цинка. Наличие двухвалентного марганца в количестве более 1 мг завышает результаты определения цинка. Наличие в пробе никеля более 0,02 мг ухудшает переход окраски индикатора, остается розовый оттенок.
Метод позволяет определить массовую долю цинка 1 мг в пробе с относительной ошибкой около 20 %.
Определение массовой доли меди
Бифторид аммония (NH 4 F HF ) или бифторид калия ( KF · HF), 20 %-ный раствор (хранится в полиэтиленовой посуде).
Йодистый калий, 40 %-ный раствор.
Титрованный раствор тиосульфата натрия 0,1-нормальной или 0,01-нормальной концентрации.
Крахмал растворимый, 0,5 %-ный свежеприготовленный раствор.
Примечание. Растворы йодистого калия и тиосульфата натрия пригодны для анализа в течение недели при условии их хранения в склянке из темного стекла и при комнатной температуре.
отбирают в коническую колбу 10 — 20 см 3 исходного раствора отложения, добавляют дистиллированной воды до 50 см 3 и доводят до рН ≈ 3 добавлением аммиака (проверяют рН по индикаторной бумаге). Затем добавляют 5 см 3 бифторида (раствор при этом обеспечивается), 10 см 3 йодистого калия, перемешивают, закрывают колбу часовым стеклом, ставят в темное место и через минуту титруют выделившийся йод тиосульфатом натрия до соломенно-желтого цвета, прибавляют в конце титрования 1 см 3 раствора крахмала и продолжают титрование до исчезновения синей окраски. Затем ставят пробу в темное место еще на 1 — 2 мин, чтобы проверить, завершилась ли реакция восстановления меди; при появлении синей окраски дотитровывают тиосульфатом натрия.
Массовую долю меди в пересчете на оксид меди ( CuO ) вычисляют по формуле, %:
где a — расход раствора тиосульфата натрия на титрование, см 3 ;
N — нормальность раствора тиосульфата натрия;
K — поправочный коэффициент к данной нормальности;
79 ,54 — эквивалентная масса CuO в данной реакции.
Примечание. Так как иодит калия бывает загрязнен иодатом ( IO 3 — ) и водные растворы йодистого калия на свету постепенно выделяют свободный йод, проводят «холостой» опыт и, если требуется, вводят соответствующую поправку.
Определение массовой доли цинка
Тиосульфат натрия, 0,1-нормальный раствор.
«Вечерний индикатор»: растворяют 0,1 г метилового оранжевого и 0,25 г индигокармина в 100 см 3 дистиллированной воды. Окраска индикатора: в щелочной среде — зеленая, при рН = 4 (переходная окраска) — серая, в более кислой — фиолетовая.
Смешанный индикатор (см. п. 2.6),
Трилон Б, 0,005-мопярный и 0,05-молярный раствор.
Сульфат меди, 0,005-молярный раствор.
в раствор, полученный после иодометрического определения содержания меди, добавляют избыток 0,5 см 3 0,1-нормального раствора тиосульфата натрия, затем капли «вечернего индикатора» и доводят до значения рН ≈ 4, добавляя аммиак до исчезновения розово-фиолетовой и появления серой или почти бесцветной (но не зеленой) окраски.
Если при определении массовой доли меди на титрование йода, выделившегося при восстановлении меди йодистым калием, пошло 5 см 3 или более 0,01-нормального (0,5 см 3 0,1-нормального) раствора тиосульфата натрия, то после доведения значения рН ≈ 4 вводят 4 — 5 капель смешанного индикатора ПАН и при интенсивном перемешивании медленно титруют раствором трилона Б до изменения окраски из фиолетово-сиреневой в желто-зеленую.
Если при определении меди пошло менее 5 см 3 0,01-нормального (0,5 см 3 0,1-нормального) раствора тиосульфата натрия, то после доведения рН ≈ 4 добавляют 1 — 2 см 3 0,005-молярного раствора сульфата меди, вводят индикатор ПАН и медленно титруют раствором трилона Б.
Массовую долю цинка в пересчете на оксид цинка ( ZnO ) вычисляют по формуле, %
где a — расход раствора трилона Б на титрование, см 3 ;
n — поправка на двухвалентную медь — 0,5 см 3 , 0,005-молярного или соответственно 0,05 см 3 0,05-молярного раствора;
M — молярность раствора трилона Б;
K — поправочный коэффициент к данной молярности;
81 ,37 — молекулярная масса ZnO в данной реакции.
Метод основан на окислении марганца до марганцовой кислоты в кислой среде персульфатом аммония в присутствии кобальтового катализатора. Образующуюся марганцовую кислоту оттитровывают щавелевокислым натрием. Для устранения присущей ионам кобальта розовой окраски, затрудняющей определение эквивалентной точки, к раствору кобальта добавляют соль меди или никеля.
Наличие хлор-ионов мешает анализу, так как они взаимодействуют с марганцовой кислотой, окисляясь до свободного хлора, и восстанавливают марганец до двухвалентного состояния, что ведет к занижению результатов анализа; их удаляют выпариванием раствора с серной кислотой.
Добавление фосфорной кислоты устраняет окраску раствора иона железа (III) и стабилизирует образующуюся марганцовую кислоту, предотвращая возможность ее разложения при кипячении раствора для разложения избытка введенного окислителя. Во избежание большого остатка персульфата аммония он вводится в анализируемый раствор небольшими порциями до полного развития окраски марганцовой кислоты.
Титрование марганцовой кислоты щавелевокислым натрием следует проводить при температуре 55 — 60 °С, так как в этом интервале температур обеспечивается достаточно быстрое их взаимодействие в эквивалентных количествах.
Метод позволяет определить массовую долю марганца 0,5 мг в пробе с относительной ошибкой около 10 %.
Серная кислота концентрированная.
Ортофосфорная кислота концентрированная.
Персульфат аммония, 20 %-ный раствор, пригоден для анализа в течение 20 дн. при условии хранения в склянках темного стекла при комнатной температуре.
Кобальтово-никелевый катализатор приготавливают растворением 1 г сернокислого кобальта (CoSO 4 · 7H 2 O) и 3 г сернокислого никеля ( NiSO 4 · 7H 2 O) в 100 см 3 воды, подкисленной 5 — 10 каплями серной кислоты.
Щавелевокислый натрий, 0,1-нормальный раствор, срок хранения приготовленного раствора не более 20 дн.
в коническую колбу на 250 см 3 отбирают 50 — 100 см 3 исходного раствора отложений, добавляют 5 см 3 серной кислоты и упаривают до объема примерно 10 см 3 для полного удаления хлор-ионов. В охлажденный раствор прибавляют 3 — 5 см 3 ортофосфорной кислоты, дистиллированную воду до объема 100 см 3 и 5 см 3 кобальтового катализатора. В интенсивно кипящий раствор вводят раствор персульфата аммония (порциями по 0,5 см 3 ) до стойкого неизменяющегося окрашивания раствора, после чего кипятят еще одну минуту. Раствор быстро охлаждают до 60 °С и при интенсивном перемешивании медленно титруют щавелевокислым натрием до исчезновения характерной малиновой окраски.
Содержание марганца в пересчете на оксид марганца ( MnO ) вычисляют по формуле, %
где а — расход раствора щавелевокислого натрия на титрование, см 3 ;
N — нормальность раствора щавелевокислого натрия;
K — поправочный коэффициент к данной нормальности;
14 ,18 эквивалентная масса MnO в данной реакции, равная пятой части молекулярной массы.
В исходный раствор отложений вводят трилон Б в избытке для образования трилонатов алюминия, железа и других комплексующихся при этом катионов. Избыток трилона Б оттитровывают раствором соли цинка в присутствии индикатора ПАН или ксиленовового оранжевого. Затем к анализируемому раствору добавляют фтористый натр. Комплекс алюминия с трилоном разрушается, алюминий связывается в более прочное соединение Na 3 AlF 6 (криолит). Освободившееся количество трилона Б, эквивалентное содержание алюминия, оттитровывают раствором соли цинка. Во избежание частичного вытеснения трилона Б из трилоната железа не следует вводить в пробу большого избытка фторида натрия.
Метод позволяет определить массовую долю алюминия 1 мг в пробе с относительной ошибкой около 10 %.
Соляная кислота, раствор 1:1.
Трилон Б, 0,025-молярный раствор.
Натрий фтористый, насыщенный раствор (4 %).
Ацетатный буферный раствор (рН = 6) : 550 г ацетата натрия CH 3 COONa · 3 H 2 O растворяют в 1 дм 3 дистиллированной воды и добавляют 100 см 3 раствора уксусной кислоты (который готовят разбавлением 58 см 3 ледяной уксусной кислоты до 1 дм 3 ).
Сернокислый цинк, 0,025-молярный раствор: 7,189 г сульфата цинка ZnSO 4 · 7H 2 O растворяют в дистиллированной воде и доводят объем до 1 дм 3 .
Поправочный коэффициент к молярности приготовленного раствора цинка устанавливают по титрованному раствору трилона Б в аммиачно-буферной среде (рН ≈ 10) с индикатором хром темно-синим.
Раствор индикатора ПАН или ксиленового оранжевого: 0,1 г индикатора ПАН растворяют в 100 см 3 этилового спирта;
0 ,13 г индикатора ксиленолового оранжевого растворяют в 2 см 3 1-нормального раствора едкого натра и разбавляют раствор водой до 100 см 3 .
отбирают в коническую колбу на 500 см 3 20 — 50 см 3 исходного раствора отложений, добавляют дистиллированной воды до 150 см 3 и доводят значение рН до 1 — 2, добавляя аммиак или соляную кислоту. Вводят 10 — 30 см 3 раствора трилона Б и медленно нейтрализуют раствором аммиака до рН ≈ 5 ÷ 6 (проверяя по индикаторной бумаге). Вводят 20 см 3 ацетатного буферного раствора, кипятят 2 — 3 мин и охлаждают в проточной воде до комнатной температуры. Добавляют 8 — 10 капель раствора индикатора ПАН или ксиленолового оранжевого и титруют окрашенную в зависимости от индикатора в желтый или оранжевый цвет пробу (избыток трилона Б) раствором сернокислого цинка до изменения цвета в фиолетово-красный. Вводят 40 см 3 раствора фтористого натрия, кипятят пробу 5 — 7 мин, охлаждают проточной водой. Добавляют 3 — 5 капель индикатора и титруют раствором сернокислого цинка окрасившуюся в желтый или оранжевый цвет пробу (если содержится алюминий) до перехода в фиолетово-красный.
Массовую долю алюминия в пересчете на оксид алюминия ( Al 2 O 3 ) вычисляют по формуле, %
где а — расход раствора цинка на второе титрование, см 3 ;
M — молярность раствора цинка;
K — поправочный коэффициент к данной нормальности;
50 ,96 — эквивалентная масса Al 2 O 3 в реакции с триионом Б, равная половине молекулярной массы.
Определению массовой доли кальция и магния (жесткости) в исходных растворах отложений титрованием трилоном Б в аммиачной буферной среде (рН = 9 ÷ 10) с хромовыми металлиндикаторами мешает наличие железа, меди, цинка, алюминия, марганца, никеля и др. Образующийся осадок гидроксида железа не титруется трилоном, но мешает отмечать изменение окраски индикатора. Ионы цинка количественно титруются совместно с кальцием и магнием. Медь и никель образуют с хромовыми индикаторами более прочные комплексы, чем кальций и магний, затягивая переход окраски индикатора в эквивалентной точке.
Мешающее влияние указанных веществ устраняется введением диэтилдитиокарбамата натрия, с которым они образуют труднорастворимые соединения: медь и никель в сильнокислой среде и до значения рН ≈ 8 ÷ 9; железо — в сильнокислой среде и до рН ≈ 5 ÷ 6; цинк и марганец — при рН ≈ 1,5 ÷ 6. Растворы диэтилдитиокарбамата натрия имеют слабощелочную реакцию. При его добавлении в пробу кислого раствора отложений значение рН повышается, проходя через интервал оптимальных значений рН ≈ 2 ÷ 3, при котором происходит образование диэтилдитиокарбаматов перечисленных мешающих веществ. Комплекс диэтилдитиокарбамата алюминия непрочный. Если массовая доля алюминия превышает 1 мг (в пробе взятой на анализ), то четкость перехода окраски индикатора ухудшается — остается розовый оттенок. Влияние алюминия устраняется введением триэтаноламина.
Исходные растворы отложений содержат обычно такие количества железа и меди, что даже при стократном и большем разбавлении дистиллированной водой добавление диэтилдитиокарбамата натрия приводит к образованию темных жидкостей, которые невозможно титровать. При этом получаются коллоидные растворы и мелкодисперсные осадки, плохо отделяемые фильтрованием. При введении же диэтилдитиокарбамата натрия в пробы исходных растворов отложений до разбавления дистиллированной водой осадки получаются более крупные, а раствор значительно прозрачнее, что дает возможность определить жесткость. Обусловлено это тем, что повышенная суммарная концентрация ионов в растворе уменьшает или полностью нейтрализует заряд коллоидных частиц и улучшает их коагуляцию. Во избежание получения заниженных результатов вследствие возможного соосаждения кальция дозировка диэтилдитиокарбамата натрия не должна превышать 0,1 г на 1 см 3 исходного раствора. Чтобы осадок получился более укрупненным, перед добавлением диэтилдитиокарбамата натрия в пробу добавляют электролит — соляную кислоту. Когда осадка много и он затрудняет наблюдение за изменением окраски раствора, его отделяют фильтрованием через беззольный бумажный фильтр или гигроскопическую вату.
Если анализируется проба с малой жесткостью, то фильтрующий материал следует предварительно промыть, как указано ниже. Даже беззольные фильтры часто содержат взаимодействующие с три лоном Б вещества в количествах, которые могут завысить результаты определения жесткости анализируемой пробы на 0,01 — 0,03 миллиграмм-эквивалента (при объеме анализируемой пробы исходного раствора 10 см 3 , общем его объеме 500 см 3 и массе навески отложения 1 г результат анализа в пересчете на массовую долю кальция будет завышен на 1 — 3 %).
Метод позволяет определить массовую долю кальция и магния 0,01 миллиграмм-эквивалента в пробе с относительной ошибкой 10 — 15 %.
Трилон Б, 0,05-молярный или 0,005-молярный раствор.
Аммиачный буферный раствор 2 %-ный NH 4 Cl и по NH 3 приготавливается смешиванием 80 см 3 25 %-ного раствора аммиака со 100 см 3 20 %-ного раствора хлористого аммония и доведением объема жидкости до 1 дм 3 дистиллированной водой.
Раствор индикатора кислотного хром темно-синего: для его приготовления растворяют 0,5 г индикатора в 20 см 3 аммиачного буферного раствора и доводят объем раствора до 100 см 3 этиловым спиртом. По данным ПО «Союзтехэнерго», хром темно-синий можно готовить только на аммиачно-буферном растворе.
Диэтилдитиокарбамат натрия кристаллический.
Соляная кислота, раствор 1:1.
Триэтаноламин, 30 %-ный раствор.
отбирают в коническую колбу 5 — 10 см 3 (если требуется, то больший объем) исходного раствора отложений, прибавляют 3 — 5 капель соляной кислоты, затем 0,3 — 0,5 г или соответственно 0,5 — 1 г кристаллического диэтилдитиокарбамата натрия. Вводить диэтилдитиокарбамат натрия лучше по частям в 3 — 4 приема. По мере его введения раствор сначала темнеет, затем осадки становятся крупнее, а раствор прозрачнее. После каждого добавления диэтилдитиокарбамата натрия пробу перемешивают, не производя резкого взбалтывания, и оставляют на 2 — 3 мин для укрупнения осадка. Когда осадок получается таким, что можно четко отличать изменение окраски, доводят объем дистиллированной водой до 100 см 3 , проверяют рН (по индикаторной бумаге) и, если требуется, Доводят его до значения 7 — 9, добавляя раствор аммиака. Затем добавляют 5 см 3 аммиачного буферного раствора, 5 — 7 капель индикатора кислотного хром темно-синего и титруют раствором трилона Б до изменения окраски из красной в сине-сиреневую. Если образовавшийся осадок диэтилдитиокарбаматов мешает четко отметить переход окраски, то после добавления диэтилдитиокарбамата натрия проверяют значение рН подученного раствора (по индикаторной бумаге) и во избежание получения заниженных результатов анализа, что наблюдается главным образом при наличии фосфатов, подкисляют раствор соляной кислотой до рН = 2 ÷ 3. Затем осадок отделяют фильтрованием раствора через беззольный фильтр или гигроскопическую вату. Если анализируют пробу с малой жесткостью, производят предварительно промывку фильтрующего материала следующим путем. Беззольный фильтр или вату помещают в воронку, как обычно для фильтрования, и промывают его 100 — 150 см 3 подкисленной до значения рН ≈ 2 ÷ 3 дистиллированной водой, подогретой до 50 — 60 °С.
Фильтр с осадком промывают несколько раз дистиллированной водой, объем фильтрата доводят до 100 см 3 и продолжают анализ, как указано вине.
Если переход окраски индикатора нечеткий, остается розовый оттенок, что может быть вызвано повышенной массовой долей алюминия, то анализ определения жесткости повторяет, добавляя в пробу перед вводом аммиачно-буферной смеси 20 см 3 30 %-ного раствора триэтаноламина. При применении триэтаноламина следует определить поправку на «холостую» пробу и, если она существенна, учесть.
Когда суммарная массовая доля кальция и магния невелика и определять отдельно кальций и магний нецелесообразно, их общую массовую долю в пересчете на оксид кальция ( СаО) вычисляют по формуле, %
где a — расход трилона Б на титрование, см 3 ;
М — молярность раствора трилона Б;
К — поправочный коэффициент к данной молярности;
56 ,08 — молекулярная масса CaO .
Примечание. При наличии фосфатов титрование трилоном Б нужно производить медленно до устойчивой сине-сиреневой окраски.
Раздельное определение массовой доли кальция и магния в одной пробе основано на том, что в сильно щелочной среде (рН ≈ 12 ÷ 13), создаваемой введением едкого натра, магний осаждается в виде гидроксида, а кальций остается в растворе и оттитровывается трилоном Б. После определения кальция введением серной или соляной кислоты понижают значение рН раствора до 8 — 9. При этом магний опять переходит в раствор. Затем добавляют аммиачный буферный раствор и определяют массовую долю магния титрованием трилоном Б. Когда общая массовая доля кальция и магния невелика, определять отдельно массовую долю кальция и магния нецелесообразно, так как это не имеет существенного значения, а результаты определения могут иметь значительную погрешность.
Чтобы устранить влияние меди, цинка, железа, марганца, никеля, в пробу исходного раствора накипи вводят диэтилдитиокарбамат натрия, как рекомендовано в п. 11. Присутствие алюминия не мешает определению массовой доли кальция. Влияние алюминия на определение массовой доли магния аналогично влиянию его при определении общей жесткости и также устраняется введением триэтаноламина (см. п. 11).
Трилон Б, 0,05-молярный или 0,005-молярный раствор.
Едкий натр, 2-нормальный раствор.
Соляная кислота, 2-нормальный раствор.
Серная кислота, 2-нормальный раствор.
Соляная кислота, раствор 1:1.
Аммиачный буферный раствор (см. п. 11).
Индикатор кислотный хром темно-синий (см. п. 11).
Диэтилдитиокарбамат натрия кристаллический.
Триэтаноламин, 30 %-ный раствор.
отбирают в коническую колбу 5 — 10 см 3 исходного раствора отложений (а если требуется, то больший объем) и обрабатывают пробу диэтилдитиокарбаматом натрия, как указано в п. 11.
Проверяют рН полученного раствора (по индикаторной бумаге), доводят его до значения 8 — 9, добавляя, если требуется, едкий натр, затем прибавляют 5 см 3 2-нормального раствора едкого натра, пробу перемешивают и выжидают около 5 мин для полного осаждения гидроксида магния. Вводят 5 — 7 капель индикатора хром темно-синего, который во избежание его адсорбции гидроксидом магния необходимо добавлять непосредственно перед титрованием. Титруют пробу раствором трилона Б до изменения окраски жидкости из розово-красной в фиолетово-голубую. Титрование заканчивают при отчетливом изменении цвета раствора, не обращая внимания на возможное последующее появление розового оттенка раствора вследствие медленного взаимодействия индикатора с гидроокисью магния. Расход трилона Б соответствует массовой доле кальция.
Для определения массовой доли магния к тому же анализируемому раствору добавляют 2-нормальную соляную кислоту, чтобы нейтрализовать введенные в пробу 5 см 3 2-нормаяьного раствора едкого натра и понизить значение рН до 8 — 9, и выжидают 5 мин для растворения гидрата окиси магния. Если присутствует магний, раствор опять приобретает красную окраску. Затем вводят 5 см 3 аммиачного раствора и титруют трилоном Б той же концентрации, что и для определения кальция, до изменения окраски анализируемого раствора в сине-сиреневую. При наличии фосфатов титрование нужно производить медленно.
Если переход окраски нечеткий и остается розовый оттенок или предыдущими анализами установлено, что в анализируемом растворе содержится алюминий в количестве, которое мешает определению магния, то перед добавлением аммиачного буферного раствора прибавляют 20 см 3 раствора триэтаноламина, затем добавляют аммиачный буферный раствор и титруют трилоном Б.
Массовую долю кальция и магния в пересчете на их оксиды в процентах вычисляют по формулам
где CaO — массовая доля кальция в отложении, %;
a 1 — расход раствора три лона Б на титрование кальция, см 3 ;
a 2 — расход раствора трилона Б на титрование магния, см 3 ;
М — молярность раствора трилона Б;
К — поправочный коэффициент к данной молярности;
40 ,32 — молекулярная масса MgO .
Определение массовой доли кальция и магния из исходного раствора накипи без отделения других компонентов, входящих в состав исходного раствора, можно выполнить с помощью фотометрического титрования на автоматическом титраторе Т-107 или Т-108.
Содержание фосфатов в исходном растворе отложения может быть определено объемным или колориметрическим методом [1 — 3]. Колориметрический метод определения фосфатов менее трудоемкий и выполняется быстрее. Поэтому в лабораториях, где имеются фотоколориметры, предпочтительно определять фосфаты колориметрическим методом.
Определение массовой доли фосфатов колориметрическим методом
Колориметрический метод определения, массовой доли фосфатов основан на образовании комплексной фосфорномолибденовой кислоты, имеющей желтую окраску, которая в присутствии восстановителей превращается в соединение синего цвета. В нижеприведенном методе восстановление производится смесью метола с метабисульфитом калия. Как для получения фосфорномолибденового комплекса, так и для его восстановления имеет существенное значение соблюдение требуемого рН. Получение фосфорномолибдата и его восстановление следует проводить в растворе серной соляной) кислоты с кислотностью до 0,6 — нормальной.
Учитывая, что исходный раствор отложений часто содержит относительно большое количество железа и меди, объем пробы исходного раствора, отбираемого для колориметрического определения фосфатов по приведенному ниже методу, ограничен 20 см 3 .
Лимонная кислота, 1-водная, квалификации «х.ч.», 10 %-ный раствор. Раствор пригоден до тех пор, пока в нем не появится осадок.
Восстанавливающий раствор, приготавливаемый из расчета: 20 г метола марки А и 120 г метабисульфита калия растворяют в 300 — 350 см 3 дистиллированной воды при температуре примерно 40 °С. Если полученный раствор мутный, его фильтруют в мерную колбу вместимостью 1 дм 3 , доливают дистиллированной водой до метки и перемешивают. Срок годности раствора не более двух недель.
Раствор молибдата аммония: к 500 см 3 дистиллированной воды добавляют 50 см 3 концентрированной серной кислоты, после охлаждения прибавляют 50 г измельченного молибдата аммония и, когда он полностью растворится, доводят объем до 1 дм 3 дистиллированной водой. Срок годности раствора не более 1 мес.
Стандартный раствор фосфата калия, содержащий 10 мг/дм 3 РO 3- 4 . Сначала готовят запасной раствор, содержащий 100 мг/дм 3 РO 3- 4 . Для этого в мерной литровой колбе растворяют в дистиллированной воде 0,143 г однозамещенного фосфата калия, предварительно выдержанного в эксикаторе над серной кислотой в течение 1 сут, доливают в колбу дистиллированной воды до метки и тщательно перемешивают.
Стандартный раствор готовят разведением запасного раствора в 10 раз. Стандартный раствор пригоден только в день приготовления.
Построение калибровочного графика:
в ряд мерных колб вместимостью 50 см 3 вводят различные количества стандартного раствора фосфата калия, содержащего 10 мг/дм 3 РO 3- 4 (например 1; 2; 3; 5; 10; 15 и 20 см 3 , что соответствует 0,01; 0,02; 0,03; 0,05; 0,10; 0,15 и 0,20 мг Р O 3- 4 .
Объем раствора в каждой колбе доводят до 40 см 3 дистиллированной водой. В одну колбу вводят только 40 см 3 дистиллированной воды (нулевая проба). Растворы нагревают на водяной бане до 40 — 60 °С, после чего в каждую колбу вводят 0,5 см 3 раствора лимонной кислоты, 2 см 3 восстанавливающего раствора и перемешивают, затем добавляют 2 см 3 раствора молибдата аммония и опять перемешивают. Спустя 10 мин добавляют в колбы дистиллированной воды до метки, растворы перемешивают и затем колориметрируют. В качестве раствора для сравнения используют дистиллированную воду со всеми реактивами. Колориметрирование проводят с красными светофильтрами при длине волны 813 нм или возможно более близкой к ней.
По результатам колориметрирования строят график, откладывая по оси абсцисс содержание РO 3- 4 в пробе (мг), а по оси ординат соответствующие показания оптической плотности.
в мерную колбу вместимостью 50 см 3 отбирают в зависимости от предполагаемого содержания фосфатов 0,5 — 20 см 3 исходного раствора отложения, добавляют дистиллированной воды до 40 см 3 нагревают на водяной бане до 40 — 60 °С. Затем вводят 0,5 см 3 раствора лимонной кислоты, 2 см 3 восстанавливающего раствора, содержимое колбы перемешивают, добавляют 2 см 3 раствора молибдата аммония и вновь перемешивают.
Спустя 10 мин доливает в колбу до метки дистиллированной воды, а затем колориметрируют.
В качестве раствора сравнения используют дистиллированную воду со всеми реактивами.
Для колориметрирования применяют те же кюветы и светофильтр, что и при построении калибровочного графика, по которому определяют содержание фосфатов в пробе.
Массовую долю фосфатов в пересчете на P 2 O 5 вычисляют по формуле, %
где a — массовая доля Р O 3- 4 в пробе, мг;
0 ,747 — коэффициент пересчета РO 3- 4 в P 2 O 5 .
Определение массовой доли фосфатов объемным методом
Сущность метода состоит в том, что фосфаты осаждают молибденовокислым аммонием из исходного раствора отложения, подкисленного азотной кислотой, в присутствии азотнокислого аммония, который прибавляют для ускорения осаждения фосфатов и уменьшения растворимости осадка (действие общего иона). Осадок фосфорномолибдата отфильтровывают, отмывают и растворяют в титрованном растворе едкого натра, добавляемого в избытке.
При этом происходит реакция, которая может быть выражена уравнением
Избыток едкого натра оттитровывают кислотой. Массовую долю фосфатов вычисляют по расходу щелочи на их растворение.
Молибденовокислый аммоний, 3 %-ный раствор.
Азотнокислый аммоний, 34 %-ный раствор.
Азотная кислота, 25 %-ный раствор.
Азотнокислый калий, 25 %-ный раствор (для промывания употребляют 1 %-ный раствор, который готовят разбавлением 25 %-ного раствора).
Едкий натр, 0,1-нормальный раствор.
Раствор соляной или серной кислоты, 0,1-нормальной концентрации.
Метилоранж, 0,1 %-ный раствор.
Фенолфталеин, 1 %-ный спиртовый раствор.
отбирают в химический стакан 25 или 50 см 3 исходного раствора отложения, приливают 20 см 3 раствора азотнокислого аммония и 10 см 3 азотной кислоты, нагревают до кипения и вливают (по стеклянной палочке) в кипящую жидкость 20 см 3 нагретого до 50 °С раствора молибденовокислого аммония. Сразу же прекращают нагревание жидкости, перемешивают ее вращением стакана (пользоваться стеклянной палочкой для перемешивания не рекомендуется) и оставляют на 2 — 3 ч. Выпавший в осадок фосфоромолибдат должен быть чисто-желтого цвета; «белесый» оттенок свидетельствует о неправильном осаждении и образовании молибденовой кислоты. В этом случае необходимо повторить осаждение, взяв новую порцию исходного раствора или растворив осадок постепенным добавлением 25 %-ного раствора аммиака, затем нагреть жидкость до кипения и добавить в нее малыми порциями 15 см 3 25 %-ного раствора азотной кислоты. После 2 — 3-часовой выдержки осадок отфильтровывают через два фильтра «синяя лента», обмывают стенки стакана и промывают осадок 1 %-ным раствором нитрата калия до нейтральной реакции промывных вод по метилоранжу.
Для проверки полноты отмывания осадка и фильтра от кислоты собирают в стаканчик или коническую колбу 10 см 3 фильтрата, вводят 1 — 2 капли метилоранжа и одну каплю 0,1-нормального раствора щелочи. Если жидкость окрасится в желтый цвет, то промывание считают законченным.
Отмытый осадок вместе с фильтром помещают в стакан, в котором проводилось осаждение, приливают 25 — 30 см 3 горячей дистиллированной воды и столько же 0,1-нормального раствора едкого натра и взбалтывают до полного растворения желтого осадка (при необходимости добавляют щелочь). После этого вводят 2 — 3 капли фенолфталеина и оттитровывают избыток щелочи раствором кислоты, титруя окрашенную в красный цвет жидкость до исчезновения окраски. Содержание фосфатов в пересчете на P 2 O 5 вычисляют по формуле, %
где ащ, ак — объемы растворов щелочи и кислоты, израсходованных при выполнении анализа, см 3 ;
N щ , Nk — нормальность растворов щелочи и кислоты;
K щ , Kk — поправочные коэффициенты растворов щелочи и кислоты к данной нормальности;
3 ,086 — эквивалентная масса P 2 O 5 в реакции со щелочью.
Массовую долю сульфатов определяют массовым или объемным методом. Сущность нижеприведенного объемного метода заключается в осаждении сульфата бария из исходного раствора, отделении и промывке осадка, растворении осадка в аммиачной среде титрованным раствором трилона Б, избыток которого оттитровывают раствором соли магния. По количеству трилона Б, израсходованного на растворение сернокислого бария, определяют содержание сульфатов в анализируемой пробе [14].
Трилон Б, 0,05-молярный раствор.
Барий хлористый, 0,05-молярный (0,1-нормальный) раствор. Для приготовления его растворяют 12,2 г BaCl 2 ´ 2 H 2 O в 1 дм 3 дистиллированной воды.
Магний хлористый, 0,05-можярный (0,1-нормальный) раствор. Приготавливают из фиксанола или 10,17 г MgCl 2 ´ 6 H 2 O растворяют в дистиллированной воде, доводят объем до 1 дм и устанавливают титр по титрованному раствору трилона.
Аммиачный буферный раствор 2 %-ный по NH 4 Cl и по NH 3 (см. п. 11).
Аммиак, 9-нормальный раствор. 67,5 см 3 концентрированного (25 %-ного) аммиака разбавляют дистиллированной водой до 100 см 3 . Раствор индикатора кислотного хром темно-синего (см. п. 11).
исходя из предполагаемого содержания сульфатов, в коническую колбу вместимостью 500 см 3 отбирают такой объем исходного раствора отложений, чтобы в нем содержалось не более 20 мг SO 2- 4 .
Для выбора объема пробы к 5 — 10 см 3 исходного раствора прибавляют 3 — 5 капель раствора хлористого бария и нагревам до кипения. При отсутствии или появлении следов белой мути отбирают 50 — 100 см 3 исходного раствора, а при значительном помутнении и выпадении осадка отбирают меньший объем пробы.
К отобранной пробе исходного раствора добавляют дистиллированной воды до 100 см 3 и подкисляют соляной кислотой до рН ≈ 1, прибавляют 10 — 15 см 3 раствора хлористого бария, кипятят 10 мин и оставляют на водяной бане или в теплом месте на 1 — 2 ч. Затем пробу фильтруют через бензольный фильтр «синяя лента», предварительно запаренный кипящей дистиллированной водой, промывают колбу с осадком несколько раз горячей дистиллированной водой, не удаляя приставшего к стенкам осадка, сливая промывные воды на фильтр с осадком. Потом фильтр с осадком промывают еще 3 — 4 раза горячей водой, проверяют на полноту промывания разбавленной серной кислотой и переносят в ту же колбу, в которой производилось осаждение, прибавляют 10 — 15 см 3 9-нормального раствора аммиака, разворачивают фильтр стеклянной палочкой на дне колбы, прибавляют 10 — 15 см 3 0,05-молярного раствора трилона Б и доводят объем до 100 см 3 , а при повышенном содержании сульфатов до 200 см 3 . При недостаточном разбавлении раствора могут получиться заниженные результаты анализа из-за неполного растворения сульфата бария, даже при значительном избытке трилона Б и поддержании требуемой щелочной среды рН ³ 10. Содержимое колбы кипятят 10 — 15 мин до полного растворения осадка, периодически проверяя (по универсальной индикаторной бумаге) рН раствора, которое должно быть не ниже 10, так как сульфат бария растворяется трилоном Б только в щелочной среде. При уменьшении рН из-за улетучивания аммиака добавляют еще 5 см 3 раствора аммиака.
После растворения осадка раствор охлаждают, вводят соответственно 5 — 10 см 3 аммиачного буферного раствора, 5 — 10 капель индикатора кислотного хром темно-синего и титруют избыток трилона Б раствором соли магния до изменения окраски из голубой в розовую.
Массовую долю сульфатов в пересчете на SO 3 в процентах вычисляют по формуле
где а T а Mg — объемы растворов трилона Б и соли магния, израсходованных при выполнении анализа, см 3 ;
M T , M Mg — молярность растворов трилона и соли магния;
KT, K Mg — поправочные коэффициенты растворов трилона и соли магния к данной молярности;
80 — молекулярная масса SO 3 в реакции с трилоном Б.
1 . Инструкция по анализу воды, пара и отложений в теплосиловом хозяйстве. — М.: Энергия, 1967.
2 . Методика контроля состояния оборудования; определение количества и состава отложений. — М.: СПО ОРГРЭС, 1976.
3 . Методика анализа накипей и отложений. — М.-Л.: Энергия, 1966.
4 . Информационное письмо № 6-82. Ускоренный анализ водорастворимой части отложений. — М.: СПО Союзтехэнерго, 1982.
5 . ГОСТ 10538.1-72. Метод определения содержания двуокиси кремния в золе.
6 . Воскресенский П.И. Техника лабораторных работ. М.: Химия, 1973.
7 . Пятницкий И.В., Симоненко В.И., Ковальчук Л.И. Ускоренные методы фотометрического определения железа в медных сплавах. Заводская лаборатория, 1982, № 3.
8 . Методика химического контроля при очистках теплообменников из латуни и медноникелевых сплавов смесью низкомолекулярных кислот и смесью их с соляной кислотой. М.: СПО Союзтехэнерго, 1980.
9 . Информационное письмо № 10-80. Ускоренное определение содержания меди, цинка и марганца в отложениях, образующихся на внутренних поверхностях нагрева теплосилового оборудования. М.: СПО Союзтехэнерго, 1980.
10 . Файнберг С.С., Филлипова А.А. Анализ руд цветных металлов. М.: Металлургиздат, 1963.
11 . Лурье Ю.Ю. Аналитическая химия промышленных сточных вод. М.: Химия, 1984.
источник