Настоящий документ устанавливает методику выполнения измерений содержаний нефтепродуктов в природных и сточных водах методом колоночной хроматографии с гравиметрическим окончанием при массовых концентрациях нефтепродуктов от 0,30 до 50,0 мг/дм 3
Мешающие влияния, обусловленные присутствием в пробе органических веществ других классов, устраняются в ходе анализа (п. 9).
ʘ Допускается использование данной методики при аварийных ситуациях для определения массовых концентраций нефтепродуктов свыше 50 мг/дм 3 . ʘ
Метод определения массовой концентрации нефтепродуктов основан на извлечении нефтепродуктов из анализируемых вод органическим растворителем, отделении от полярных соединений других классов колоночной хроматографией на оксиде алюминия и количественном определении гравиметрическим методом.
Настоящая методика обеспечивает получение результатов анализа с погрешностью, не превышающей значений, приведенных в таблице 1.
Диапазон измерений, значения показателей точности, повторяемости и воспроизводимости
Показатель точности (границы относительной погрешности при вероятности Р = 0,95), ± d , %
(относительное сред- неквадратическое отклонение повторяемости) s г , %
(относительное среднеквадратическое отклонение воспроизводимости), s R , %
Значения показателя точности методики используют при:
— оформлении результатов анализа, выдаваемых лабораторией;
— оценке деятельности лабораторий на качество проведения испытаний;
— оценке возможности использования результатов анализа при реализации методики в конкретной лаборатории.
При выполнении измерений должны быть применены следующие средства измерений, оборудование и материалы:
3.1. Средства измерений, вспомогательное оборудование
Весы лабораторные, 2 класса точности, ГОСТ 24104
Вентилятор комнатный типа ВН10-УЧ, ГОСТ 7402
Термометр КШ-14/23, ТУ 25-2021.007-88
Стаканчики для взвешивания (бюксы), ГОСТ 25336
Пипетки мерные с делениями 0,1 см 3 4(5)-2-1(2);
Колонка с оксидом алюминия
Бутыли из стекла с притертыми пробками вместимостью 2000 — 3000 см 3 для отбора и хранения проб
Алюминии оксид, ТУ 6-09-3916
Бумага индикаторная универсальная, ТУ 6-09-1181 ʘ
При выполнении измерений массовой концентрации нефтепродуктов в пробах природных и сточных вод соблюдают следующие требования безопасности:
4.1. При выполнении анализов необходимо соблюдать требования техники безопасности при работе с химическими реактивами по ГОСТ 12.1.007.
4.2. Электробезопасность при работе с электроустановками по ГОСТ 12.1.019.
4.3. Организация обучения работающих безопасности труда по ГОСТ 12.0.004.
4.4. Помещение лаборатории должно соответствовать требованиям пожарной безопасности по ГОСТ 12.1.004 и иметь средства пожаротушения по ГОСТ 12.4.009.
Выполнение измерений может производить химик-аналитик, владеющий техникой гравиметрического метода анализа.
При выполнении измерений соблюдают следующие условия:
температура окружающего воздуха (20 ± 5) ℃ ;
атмосферное давление (84 — 106) кПа (630 — 800 мм.рт.ст);
относительная влажность (80 ± 5) %;
частота переменного тока (50 ± 1) Гц;
напряжение в сети (220 ± 10) В.
Отбор проб производится в соответствии с требованиями ГОСТ Р 51592-2000 «Вода. Общие требования к отбору проб» ʘ .
7.1. Пробы воды для параллельных определений отбирают в отдельные стеклянные емкости с притертыми пробками. Пробу для одного определения используют полностью. Если определение нефтепродуктов в день отбора невозможно, то пробы консервируют 2 — 4 см 3 экстрагента (четыреххлористый углерод, хлороформ) на 1 дм 3 воды. Законсервированные пробы могут храниться в течение двух недель.
При определении нефтепродуктов методом колоночной хроматографии с гравиметрическим окончанием объем пробы (при концентрации нефтепродуктов 0,3 — 3,0 мг/дм 3 ) должен составлять не менее 3 — 3,5 дм 3 .
7.2. При отборе проб составляется сопроводительный документ по утвержденной форме, в котором указывается:
— цель анализа, предполагаемые загрязнители;
— должность, фамилия отбирающего пробу, дата.
При подготовке к выполнению измерений проводят следующие работы:
ʘ 8.1. Подготовка оксида алюминия II степени активности ʘ
Реактив перед употреблением прокаливают в муфельной печи при 600 °С в течение 4 часов, дают остыть в эксикаторе и добавляют 3 % (по массе) дистиллированной воды. Хранят в склянке с притертой пробкой.
ʘ 8.2. Подготовка натрия сернокислого безводного ʘ
Перед использованием реактив прокаливают в сушильном шкафу при температуре 105 °С в течение 3 часов.
ʘ 8.3. Подготовка колонки с оксидом алюминия ʘ
Колонка с оксидом алюминия представляет собой стеклянную трубку длиной 10 см и диаметром 0,7 — 1,0 см с оттянутым нижним концом до диаметра 0,1 см. В трубку помещают стеклянную вату слоем 0,5 см, затем 6 г оксида алюминия и снова стеклянную вату. В качестве колонки можно использовать обычную пипетку, градуированную на 10 см 3 . Оксид алюминия в колонке меняют после каждой пробы. Использованный оксид алюминия можно регенерировать промыванием его четыреххлористым углеродом или хлороформом, испарением растворителя и последующим его прокаливанием.
Мешающие влияния, обусловленные присутствием в пробе органических веществ других классов, устраняются в ходе анализа: одни остаются нерастворимыми в гексане, другие (фенолы, нафтеновые кислоты) сорбируются оксидом алюминия.
10.1. Определение при концентрации нефтепродуктов 0,3 — 3,0 мг/дм 3
При выполнении измерений массовой концентрации нефтепродуктов в пробах природных и сточных вод выполняют следующие операции:
3 — 3,5 дм 3 исследуемой пробы воды подкисляют соляной кислотой (плотн. 1,19 г/см 3 ) до рН 3 хлороформа или четыреххлористого углерода, погружают мешалку так, чтобы лопасти её были в воде на 50 мм выше границы слоев воды и растворителя и перемешивают в течение 10 мин.

Затем переносят большую часть водного слоя в другой сосуд такой же вместимости, а оставшийся водный слой и слой хлороформа помещают в делительную воронку вместимостью 500 — 700 см 3 .
Через 15 минут сливают нижний слой хлороформа в коническую колбу (Эрленмейера) вместимостью 500 см 3 , стараясь не захватить при этом ни воды, ни промежуточного слоя эмульсии.
Переливают водный раствор из второго сосуда снова в первый, туда же переносят оставшийся в деятельной воронке водный слой с эмульсией, добавляют вторую порцию хлороформа 150 см 3 и снова перемешивают мешалкой в течение 5 — 7 мин. Снова сливают большую часть водного слоя, остаток переносят в ту же делительную воронку.
Через 15 мин отделяют второй экстракт и присоединяют его к первому, не захватывая при этом водного слоя. Затем небольшим количеством хлороформа (около 50 см 3 ) обмывают стенки сосуда, в котором проба находилась до экстракции, переносят в ту же делительную воронку, взбалтывают, дают некоторое время отстояться и присоединяют слой хлороформа к первым двум экстрактам.
В проведении третьей экстракции обычно нет необходимости.
Экстракцию хлороформом можно также проводить следующим способом: в делительную воронку вместимостью 1 — 2 дм 3 помещают 3 раза по 1 дм 3 исследуемой воды и последовательно взбалтывают с двумя порциями по 20 см 3 хлороформа. Таким образом, на экстракцию из 3 дм 3 анализируемой пробы будет израсходовано 120 см 3 хлороформа. Экстракты соединяют, прибавляют к ним 50 см хлороформного раствора, полученного при ополаскивании сосуда, где хранилась проба (*) .
(*) Склянку, в которой находилась проба, ополаскивают растворителем, который используется для экстракции.
Колбу с экстрактом присоединяют к холодильнику, помещают её в кипящую водяную баню или ставят на горячую закрытую плитку и отгоняют хлороформ до тех пор, пока в колбе не останется 10 — 20 см 3 раствора. Дают колбе остыть и разбирают прибор.
Остатки хлороформа удаляют при комнатной температуре. Предварительно взвешенный бюкс (с крышкой) помещают в вытяжном шкафу на расстоянии 25 — 35 см от обычного комнатного вентилятора, снимают крышку, заполняют бюкс на три четверти полученным экстрактом, включают вентилятор; по мере испарения экстракт подливают в бюкс, пока не перенесут полностью. Колбу из-под экстракта обмывают небольшой порцией хлороформа и переносят в тот же бюкс.
Когда в бюксе останется менее 0,5 см 3 хлороформного раствора, выключают вентилятор и продолжают испарение на воздухе, взвешивая бюкс каждые 2 мин. Перед взвешиванием его закрывают крышкой и вновь снимают крышку для дальнейшего испарения. Когда масса перестанет изменяться, испарение заканчивают.
Разность между массой бюкса с остатком после удаления хлороформа и массой пустого бюкса показывает общее содержание экстрагируемых хлороформом веществ.
Остаток после отгонки хлороформа растворяют в 1 — 2 см 3 предварительно высушенного сульфатом натрия н-гексана или петролейного эфира. Полученный раствор вместе с частицами нерастворившегося остатка, если такие окажутся, переносят в колонку с оксидом алюминия, под которую подставляют чистую сухую колбу. Бюкс несколько раз обмывают маленькими порциями н-гексана, переносят каждую порцию в колонку с оксидом алюминия. Колонку промывают еще несколькими порциями н-гексана (всего 40 — 45 см 3 ), собирая их в ту же колбу. Не следует при этом допускать, чтобы уровень н-гексана в колонке опускался ниже верхней границы слоя оксида алюминия.
Из полученного раствора нефтепродуктов в н-гексане, свободном от полярных соединений, удаляют н-гексан, испаряя его из бюкса при комнатной температуре вентилятором так же, как удаляли раньше хлороформ. Разность между массой бюкса с остатком после удаления н-гексана и массой пустого бюкса показывает содержание нефтепродуктов во взятом для исследования объеме пробы.
10.2. Определение нефтепродуктов в концентрациях выше 3,0 мг/дм 3
Определение проводят так же, как описано в п. 10.1, но только с меньшим объемом исследуемой воды. Берут для анализа 100 — 1000 см 3 воды, соответственно взятому объему воды уменьшают и количество применяемого для экстракции растворителя.
Содержание массовой концентрации нефтепродуктов X (мг/дм 3 ) рассчитывают по формуле:
где m1, — масса бюкса с остатком после удаления гексана, мг,
m2 — масса пустого бюкса, мг;
V — объем пробы, взятой для анализа, см 3 .
За результат анализа Хср принимают среднее арифметическое значение двух параллельных определений Х1 и Х2
для которых выполняется следующее условие:
где r — предел повторяемости, значения которого приведены в таблице 2.
Значения предела повторяемости при вероятности Р = 0,95
Диапазон измерений, мг/дм 3
Предел повторяемости (относительное значение допускаемого расхождения между двумя параллельными результатами измерений), г, %
При невыполнении условия (1) могут быть использованы методы проверки приемлемости результатов параллельных определений и установления окончательного результата согласно раздела 5 ГОСТ Р ИСО 5725-6.
Расхождение между результатами анализа, полученными в двух лабораториях, не должно превышать предела воспроизводимости. При выполнении этого условия приемлемы оба результата анализа, и в качестве окончательного может быть использовано их среднее арифметическое значение. Значения предела воспроизводимости приведены в таблице 3.
Значения предела воспроизводимое при вероятности Р = 0,95
Диапазон измерений, мг/дм 3
Предел воспроизводимости (относительное значение допускаемого расхождения между двумя результатами измерений, полученными в разных лабораториях), R, %
источник
ПНД Ф 14.1:2:3.110-97 Количественный химический анализ вод. Методика измерений массовой концентрации взвешенных веществ в пробах природных и сточных вод гравиметрическим методом / 14 1 2 3 110 97
ФЕДЕРАЛЬНАЯ СЛУЖБА ПО НАДЗОРУ
В СФЕРЕ ПРИРОДОПОЛЬЗОВАНИЯ
«Федеральный центр анализа и
оценки Техногенного
воздействия»
________________ В.В. Новиков
КОЛИЧЕСТВЕННЫЙ ХИМИЧЕСКИЙ АНАЛИЗ ВОД
МЕТОДИКА ИЗМЕРЕНИЙ
МАССОВОЙ КОНЦЕНТРАЦИИ ВЗВЕШЕННЫХ ВЕЩЕСТВ
В ПРОБАХ ПРИРОДНЫХ И СТОЧНЫХ ВОД
ГРАВИМЕТРИЧЕСКИМ МЕТОДОМ
Методика допущена для целей государственного
экологического контроля
Методика измерений аттестована Центром метрологии и сертификации «СЕРТИМЕТ» Уральского отделения РАН (Аттестат аккредитации № RA.RU.310657 от 12.05.2015), рассмотрена и одобрена федеральным государственным бюджетным учреждением «Федеральный центр анализа и оценки техногенного воздействия» (ФГБУ «ФЦАО»).
Настоящее издание методики введено в действие взамен предыдущего издания ПНД Ф 14.1:2.110-97 и действует с 01 декабря 2016 года до выхода нового издания.
Сведения об аттестованной методике измерений переданы в Федеральный информационный фонд по обеспечению единства измерений.
Заместитель директора ФГБУ «ФЦАО»
Разработчик: © ООО НПП «Акватест»
Настоящий нормативный документ устанавливает методику измерений массовой концентрации взвешенных веществ в диапазоне от 3,0 до 5000 мг/дм 3 в пробах природных (поверхностных и подземных) и сточных (производственных, хозяйственно-бытовых, ливневых, очищенных) вод гравиметрическим методом.
Результаты измерений могут быть некорректными при наличии в пробе значительных количеств нефтепродуктов и жиров, поэтому при отборе пробы не допускают попадания в нее поверхностной пленки, а также плавающих частиц (кусочков бумаги, листьев, травы и т.п.).
ГОСТ 12.0.004-90 ССБТ. Организация обучения безопасности труда. Общие положения.
ГОСТ 12.1.004-91 ССБТ. Пожарная безопасность. Общие требования.
ГОСТ 12.1.005-88 ССБТ. Общие санитарно-гигиенические требования к воздуху рабочей зоны.
ГОСТ 12.1.007-76 ССБТ. Вредные вещества. Классификация и общие требования безопасности.
ГОСТ 12.1.009-83 ССБТ. Пожарная техника для защиты объектов. Основные виды. Размещение и обслуживание.
ГОСТ Р 12.1.019-2009 ССБТ. Электробезопасность. Общие требования и номенклатура видов защиты.
ГОСТ 17.1.5.04-81 Охрана природы. Гидросфера. Приборы и устройства для отбора, первичной обработки и хранения проб природных вод. Общие технические условия.
ГОСТ 17.1.5.05-85 Охрана природы. Гидросфера. Общие требования к отбору проб поверхностных и морских вод, льда и атмосферных осадков.
ГОСТ 1770-74 Посуда мерная лабораторная стеклянная. Цилиндры, мензурки, колбы, пробирки. Технические условия.
ГОСТ 3118-77 Реактивы. Кислота соляная. Технические условия.
ГОСТ 3145-84 Часы механические с сигнальным устройством. Общие технические условия
ГОСТ 3956-76 Силикагель технический. Технические условия.
ГОСТ 6709-72 Вода дистиллированная. Технические условия.
ГОСТ 9147-80 Посуда и оборудование лабораторные фарфоровые. Технические условия.
ГОСТ 14919-83 Электроплиты, электроплитки и жарочные электрошкафы бытовые. Общие технические условия.
ГОСТ 21241-89 (СТ СЭВ 5204-85) Пинцеты медицинские. Общие технические требования и методы испытаний.
ГОСТ 25336-82 Посуда и оборудование лабораторные стеклянные. Типы. Основные параметры и размеры.
ГОСТ 27384-2002 Вода. Нормы погрешности измерений показателей
ГОСТ 31861-2012 Вода. Общие требования к отбору проб.
ГОСТ Р 53228-2008 Весы неавтоматического действия. Часть 1. Метрологические и технические требования. Испытания.
ГОСТ OIML R 76-1-2011 ГСИ Государственная система обеспечения единства измерений. Весы неавтоматического действия. Часть 1. Метрологические и технические требования. Испытания.
ГОСТ Р ИСО 5725-6-2002 Точность (правильность и прецизионность) методов и результатов измерений. Часть 6. Использование значений точности на практике.
ТУ 6-09-1678-95 Фильтры обеззоленные (белая, красная, синяя ленты).
ТУ 6-09-4711-81 Реактивы. Кальций хлористый (обезвоженный), чистый. Технические условия.
ТУ 64-1-909-80 Шкафы сушильно-стерилизационные ШСС-80П.
ТУ 2265-011-43153636-2015 Мембрана ацетатцеллюлозная Владипор МФАС-ОС-2-37 мм (0,45 мкм).
ТУ 3616-001-32953279-97 Приборы вакуумного фильтрования ПВФ-35 и ПВФ-47.
Гравиметрический метод измерения массовой концентрации взвешенных веществ основан на выделении их из пробы фильтрованием воды через мембранный фильтр с диаметром пор 0,45 мкм или бумажный фильтр «синяя лента» и взвешивании осадка на фильтре после высушивания его при (105 ± 2) °С до постоянной массы.
4.1 Настоящая методика обеспечивает получение результатов измерений с погрешностями, не превышающими значений, приведённых в таблице 1.
Значения показателя точности методики используют при:
— оформлении результатов измерений, выдаваемых лабораторией;
— оценке деятельности лабораторий на качество проведения испытаний;
— оценке возможности использования результатов измерений при реализации методики в конкретной лаборатории.
Диапазон измерений массовой концентрации взвешенных веществ, мг/дм 3
Показатель точности (границы относительной погрешности при вероятности Р = 0,95), ±δ, %
Показатель повторяемости (относительное среднеквадратическое отклонение повторяемости), σ r , %
Показатель воспроизводимости (относительное среднеквадратическое отклонение воспроизводимости), σ R , %
Весы лабораторные общего назначения специального или высокого класса точности с наибольшим пределом взвешивания 210 г
Цилиндры мерные исполнения 1, 3 вместимостью 25, 50, 100, 250, 500 и 1000 см 3
Часы механические с сигнальным устройством
Воронки лабораторные диаметром 75, 100 и 150 мм
Стакан В-1, ТХС вместимостью 500 см 3
Стаканчики для взвешивания (бюксы) низкие СН-45/13 или СН-60/14
Чашки биологические низкие (Петри) диаметром 100 — 150 мм
Шкаф сушильный общелабораторного назначения, обеспечивающий поддержание температуры нагрева (105 ± 2)°С
Электроплитка с закрытой спиралью и регулируемой мощностью нагрева
Прибор вакуумного фильтрования ПВФ-35 или ПВФ-47
Склянки для хранения проб вместимостью 500, 1000 и 2000 см 3 или
Бутыли полиэтиленовые (полипропиленовые) для хранения проб вместимостью 500, 1000 и 2000 см 3
Средства измерений должны быть поверены в установленные сроки.
Допускается использование других, в том числе импортных, средств измерений утвержденных типов и вспомогательных устройств с характеристиками не ниже указанных в п. 5.1.
Фильтры мембранные Владипор типа МФАС-ОС-2 (0,45 мкм) с диаметром 37 или 47 мм или
Фильтры бумажные обеззоленные «синяя лента» диаметром 90 или 110 мм
Хлорид кальция безводный (для эксикатора) или
Допускается использование реактивов и материалов, изготовленных по другой нормативно-технической документации, в том числе импортных, с характеристиками не ниже указанных в п. 5.2.
6.1. При выполнении измерений необходимо соблюдать требования техники безопасности при работе с химическими реактивами по ГОСТ 12.1.007.
6.2. Электробезопасность при работе с электроустановками обеспечивается по ГОСТ Р 12.1.019.
6.3. Организация обучения работающих безопасности труда проводится по ГОСТ 12.0.004.
6.4. Помещение лаборатории должно соответствовать требованиям пожарной безопасности по ГОСТ 12.1.004 и иметь средства пожаротушения по ГОСТ 12.4.009.
6.5. Содержание вредных веществ в воздухе помещения лаборатории не должно превышать установленных предельно допустимых концентраций в соответствии с ГОСТ 12.1.005.
К выполнению измерений и обработке их результатов допускаются лица, имеющие квалификацию техника-химика или лаборанта-химика и владеющие техникой гравиметрического анализа.
При выполнении измерений в лаборатории должны быть соблюдены следующие условия:
— температура окружающего воздуха (22 ± 6) °С;
— атмосферное давление (84 — 106) кПа;
— относительная влажность не более 80 % при температуре 25 °С;
— частота переменного тока (50 ± 1) Гц;
— напряжение в сети (220 ± 22) В.
9.1. Отбор проб для выполнения измерений массовой концентрации взвешенных веществ производится в соответствии с ГОСТ 31861 и ГОСТ 17.1.5.05.
9.2. Оборудование для отбора проб должно соответствовать ГОСТ 31861, ГОСТ 17.1.5.04 и ГОСТ 17.1.5.05.
9.3. Пробы отбирают в стеклянную или пластиковую посуду, предварительно промытую раствором соляной кислоты, а затем дистиллированной водой. При отборе посуду ополаскивают отбираемой водой.
9.4. Объем отбираемой пробы должен быть не менее 1000 см 3 при массовой концентрации взвешенных веществ ниже 50 мг/дм 3 и не менее 500 см 3 при массовой концентрации взвешенных веществ выше 50 мг/дм 3 .
9.5. Пробу анализируют как можно скорее, но не позднее 24 ч после отбора.
9.6. При отборе проб составляется сопроводительный документ по утвержденной форме, в котором указывается:
— место, дата и время отбора;
— должность, фамилия сотрудника, отбирающего пробу.
Фильтры кипятят в дистиллированной воде 5 — 10 мин. Кипячение проводят 3 раза, сливая после каждого раза воду и заменяя ее свежей. Затем фильтры помещают в чашки Петри, подсушивают на воздухе в течение 25 — 30 мин и сушат в сушильном шкафу при (105 ± 2) °С в течение 1 ч. Чистые фильтры хранят в закрытых чашках Петри.
Непосредственно перед использованием фильтры маркируют карандашом с мягким грифелем, с помощью пинцета помещают в маркированные бюксы, сушат при (105 ± 2) °С в течение 1 ч, охлаждают в эксикаторе и, закрыв бюксы крышками, взвешивают. Повторяют процедуру сушки до тех пор, пока разница между взвешиваниями будет не более 0,5 мг.
Бумажные обеззоленные фильтры «синяя лента» маркируют, складывают, помещают в воронки и промывают 150 — 200 см 3 дистиллированной воды. Затем пинцетом вынимают фильтр из воронки, складывают, помещают в маркированные бюксы и высушивают в сушильном шкафу при (105 ± 2) °С в течение 2 ч. Охлаждают бюксы с фильтрами в эксикаторе и, закрыв их крышками, взвешивают. Повторяют процедуру сушки до тех пор, пока разница между взвешиваниями будет не более 0,5 мг.
По готовности фильтра выполняют измерения в соответствии с п. 12.2. Если невозможно выполнить измерения сразу после подготовки фильтра, его хранят в закрытом бюксе в эксикаторе или в закрытой емкости, исключающей попадание пыли на поверхность бюкса.
30 см 3 соляной кислоты смешивают с 170 см 3 дистиллированной воды. Раствор хранят в плотно закрытой посуде не более 1 года.
Подготовку прибора для вакуумного фильтрования осуществляют в соответствии с инструкцией по его эксплуатации.
Подготовленный и взвешенный мембранный фильтр пинцетом извлекают из бюкса и закрепляют в ячейке прибора вакуумного фильтрования. Затем анализируемую пробу воды тщательно перемешивают энергичным взбалтыванием и переливают нужный для фильтрования объем в мерный цилиндр. Этот объем зависит от содержания взвешенных веществ в воде и подбирается с таким расчетом, чтобы масса осадка взвешенных веществ на фильтре была не менее 3 мг и не превышала 250 мг. Рекомендуемые объемы пробы для фильтрования приведены в таблице 2.
Предполагаемый диапазон массовой концентрации взвешенных веществ, мг/дм 3
Отбираемый для фильтрования объем пробы воды, см 3
После пропускания пробы воды через фильтр ополаскивают мерный цилиндр дважды 4 — 5 см 3 дистиллированной воды, переносят смывы на фильтр, а приставший к стенкам ячейки для фильтрования осадок дважды смывают фильтратом порциями по 10 см 3 на фильтр.
Фильтр с осадком извлекают пинцетом из устройства для фильтрования, помещают в тот же бюкс, в котором его взвешивали до фильтрования, подсушивают сначала 15 — 20 мин на воздухе, а затем в сушильном шкафу при (105 ± 2) °С в течение 1 ч со снятой крышкой. Крышка бюкса должна находиться возле бюкса. После этого бюкс охлаждают в эксикаторе, закрывают крышкой и взвешивают.
Повторяют процедуру сушки до тех пор, пока разница между взвешиваниями будет не более 0,5 мг при массе осадка до 50 мг и 1 мг при массе более 50 мг.
Использование бумажных фильтров допускается в случае отсутствия в лаборатории устройства для вакуумного фильтрования с мембранным фильтром. В этом случае в рабочем журнале указывается, что результат измерений получен с использованием бумажного фильтра.
Подготовленный бумажный фильтр помещают в воронку, смачивают небольшим количеством дистиллированной воды для хорошего прилипания и пропускают отмеренный объем тщательно перемешанной анализируемой пробы воды, подобранный с таким расчетом, чтобы масса осадка взвешенных веществ на фильтре находилась в пределах от 3 до 250 мг (таблица 2).
После пропускания пробы воды через фильтр ополаскивают мерный цилиндр дважды 4 — 5 см 3 дистиллированной воды, перенося смывы на фильтр. Промывают фильтр 10 см 3 дистиллированной воды, дают воде полностью стечь, пинцетом осторожно вынимают фильтр с осадком и помещают в тот же бюкс, в котором его взвешивали до фильтрования. Фильтр высушивают 2 ч при (105 ± 2) °С, охлаждают в эксикаторе и, закрыв бюкс крышкой, взвешивают.
Повторяют процедуру сушки, пока разница между взвешиваниями будет не более 0,5 мг при массе осадка до 50 мг и 1 мг при массе более 50 мг.
Массовую концентрацию взвешенных веществ в анализируемой пробе воды X, мг/дм 3 , рассчитывают по формуле:
где mфо — масса бюкса с мембранным или бумажным фильтром с осадком взвешенных веществ, г;
mф — масса бюкса с мембранным или бумажным фильтром без осадка, г;
V — объем профильтрованной пробы воды, дм 3 .
Расхождение между результатами измерений, полученными в условиях воспроизводимости, не должно превышать предела воспроизводимости (таблица 3).
Диапазон измерений массовой концентрации взвешенных веществ, мг/дм 3
Предел повторяемости (относительное значение допускаемого расхождения между двумя результатами параллельных измерений), r,%
Предел воспроизводимости (относительное значение допускаемого расхождения между двумя результатами измерений, полученными в разных лабораториях), R, %
источник
Методы анализа воды: гравиметрические, титриметрические, фотометрические, потенциометрические, вольтамперометрические.
Гравиметрический – основан на определении массы вещества. В ходе анализа вещество отгоняется в виде какого-либо летучего соединения или осаждается из раствора в виде малорастворимого соединения. Осадок взвешивается в виде соединения строго определенного состава, весовая форма по составу совпадает с осаждаемой. По весу высушенного или прокаленного осадка вычисляется содержание определенного компонента в данном образце. Достоинства: высокая точность, отсутствие необходимости калибровки, простота. Недостатки: значительный расход времени на выполнение анализа.
Титриметрический. Основан на точном измерении количества реактива израсходованного на реакцию с определенными веществами. Титрированный раствор – раствор, концентрация которого известна с высокой точностью. Титрование – прибавление титрованного раствора к анализируемому для точного определения эквивалентного количества. Момент титрирования – точка эквивалентности. Титрирующий раствор – титрант. Используются реакции кислотно-основного взаимодействия, удовлетворяющие требованиям, которые предъявляются к титриметрическим реакциям. Взаимодействие должно происходить полностью и с высокой скоростью. Достоинства: быстрота выполнения, простота оборудования, удобство выполнения серийных анализов, большой набор химических реакций. Недостатки: необходимость предварительной стандартофикации растворов титранта и калибровки мерной посуды.
Фотометрический. Измеряет поглощение света анализируемым раствором обычно после введения в него реактива, реагирующего с определенным компонентом сточной воды с образованием интенсивно поглощающего свет соединения. Приборы: Источник света – светофильтр – кювета с раствором – детектор. Конструкция прибора зависит от области спектра применения. Излучение выбирают такое, что бы соединение имело max светопоглощение, а примеси – min. Достоинства – широкая область применения, высокая чувствительность. Недостатки: калибровка аппаратуры, посуды.
Потенциометрия и потенциометрическое титрование. Потенциометрия основана на измерении небольших равновесных напряжений между электродами гальванической ячейки. Метод можно применять для установления активности веществ в растворе (прямая потенциометрия) и для нахождения точки эквивалентности при титриметрических определениях (потенциометрическое титрование). Прямая ПМ находит применение при определении рН растворов, а также многих ионов с использованием ионоселективных электродов. В анализе природных вод и питьевой воды ионоселективные электроды применяют для определения кадмия, меди, свинца, серебра, щелочных металлов, бромид-, хлорид-, цианид-, фторид-, иодид — и сульфид-ионов.
Вольтамперометрические методы анализа. Это совокупность методов исследования кривых ток-потенциал и их зависимостей от электродных реакций и концентраций определяемых веществ. Один из основных ВАМ методов – полярография. Метод заключается в получении и анализе кривых ток-потенциал на ртутном капельном электроде. Методом полярографии можно определить любые вещества, способные к эл-хим превращениям на электродах. Качественная информация следует из значения потенциала полуволны (φ1/2), количественная – из определения высоты волны (id).
Типичная полярографическая волна, используемая для качественного и количественного определения электродно-активных веществ.
47. Контроль и управление качеством воды в водных объектах:
а) ПДК загрязняющих веществ;
б) предельно-допустимая нагрузка на водный объект (ПДН), чем она определяется; в) предельно-допустимый сброс (ПДС). Понятие о ХПК и БПК.
а) для водной среды ПДК загрязняющих веществ означает такую концентрацию вещества выше которой вода становится неприродной для одного или нескольких видов водопользования.
б) степень предельно допустимого загрязнения воды определяется его физическими особенностями (температурой, скоростью течения), а так же способность к нейтрализации примеси, есть предельно допустимая нагрузка на водный объект ПДН, так как использование воды связано с её изъятием, а значит с угрозой истощения водного объекта, разрушение его экологической системы.
в) Сброс сточных вод должен осуществляться до уровня саморазгрузки. Либо сточные воды должны очищаться или разбавляться перед сбросом, либо рассеиваться сразу после сброса до установления норматива.
ПДС – устанавливается санэпидем службой для каждого предприятия с учетом:
б) осимиллирующие способности водного объекта
в) сброс других производств
д) с учетом хим. состава и рельефа.
ХПК – хим. потребл. кислорода. Определяется как количество кислорода потребляемого при хим. окислении под воздействием окислителей, содержащихся в воде органич и неорганич веществ.
БПК – биологическая потребляемость кислорода. Это количество кислорода, израсходованного за определенный промежуток времени (5, 20 суток) на аэробное биохимич окисление, то есть на разложение органич соединений микроорганизмами.
Защита гидросферы.
1) Развитие безотходных и безводных технологий, систем замкнутого водоснабжения
3) очистка и обеззараживание поверхностных вод, использующихся для водоснабжения.
4) закачка сточных вод в глубокие водоносные горизонты.
Организация стока поверхностных вод: Наибольшее количество влаги на земном шаре испаряется с поверхности морей и океанов (88‰).
Поперечные профили набережных и береговой полосы: На городских территориях берегоукрепление проектируют с учетом технических и экономических требований, но особое значение придают эстетическим.
Папиллярные узоры пальцев рук — маркер спортивных способностей: дерматоглифические признаки формируются на 3-5 месяце беременности, не изменяются в течение жизни.
Опора деревянной одностоечной и способы укрепление угловых опор: Опоры ВЛ — конструкции, предназначенные для поддерживания проводов на необходимой высоте над землей, водой.
источник
ПНД Ф 14.1:2.122-97
Количественный химический анализ вод. Методика измерений массовой концентрации жиров в поверхностных и сточных водах гравиметрическим методом
Купить ПНД Ф 14.1:2.122-97 — бумажный документ с голограммой и синими печатями. подробнее
Распространяем нормативную документацию с 1999 года. Пробиваем чеки, платим налоги, принимаем к оплате все законные формы платежей без дополнительных процентов. Наши клиенты защищены Законом. ООО «ЦНТИ Нормоконтроль»
Наши цены ниже, чем в других местах, потому что мы работаем напрямую с поставщиками документов.
- Срочная курьерская доставка (1-3 дня)
- Курьерская доставка (7 дней)
- Самовывоз из московского офиса
- Почта РФ
Документ устанавливает методику измерений массовой концентрации жиров в поверхностных и сточных водах гравиметрическим методом. Диапазон измерений от 0,5 до 50 мг/дм3.
2. Приписанные характеристики показателей точности измерений
3. Средства измерений, вспомогательные оборудование, реактивы и материалы
3.1 Средства измерений и вспомогательное оборудование
5. Требования безопасности, охраны окружающей среды
6. Требования к квалификации операторов
7. Требования к условиям измерений
8. Подготовка к выполнению измерений
8.1 Подготовка посуды для отбора проб
8.2 Отбор и хранение проб воды
8.3 Подготовка оксида алюминия II степени активности
10. Обработка результатов измерений
11. Оформление результатов измерений
12. Контроль точности результатов измерений
12.2 Оперативный контроль процедуры измерений с использованием образцов для контроля
13. Проверка приемлемости результатов, полученных в двух лабораториях
Приложение А (информационное). Бюджет неопределенности измерений
Чтобы бесплатно скачать этот документ в формате PDF, поддержите наш сайт и нажмите кнопку:
ФЕДЕРАЛЬНАЯ СЛУЖБА ПО НАДЗОРУ
В СФЕРЕ ПРИРОДОПОЛЬЗОВАНИЯ
И.о. директора ФБУ «Федеральный
центр анализа и оценки техногенного
_________________ С.А. Хахалин
КОЛИЧЕСТВЕННЫЙ ХИМИЧЕСКИЙ АНАЛИЗ ВОД
МЕТОДИКА ИЗМЕРЕНИЙ МАССОВОЙ
КОНЦЕНТРАЦИИ ЖИРОВ В ПОВЕРХНОСТНЫХ
И СТОЧНЫХ ВОДАХ
ГРАВИМЕТРИЧЕСКИМ МЕТОДОМ
Методика допущена для целей государственного
экологического контроля
МОСКВА 1997 г.
(издание 2011 г.)
Методика рассмотрена и одобрена федеральным бюджетным учреждением «Федеральный центр анализа и оценки техногенного воздействия (ФБУ «ФЦАО»).
Главный инженер ФБУ «ФЦАО», к.х.н.
«Федеральный центр анализа и оценки техногенного воздействия» (ФБУ «ФЦАО»)
Настоящий документ устанавливает методику измерений массовой концентрации жиров в поверхностных и сточных водах гравиметрическим методом.
Диапазон измерений от 0,5 до 50 мг/дм 3 .
Значения показателя точности измерений 1 — расширенной относительной неопределенности измерений по настоящей методике при коэффициенте охвата 2 приведены в таблице 1. Бюджет неопределенности измерений приведен в Приложении А
Таблица 1 — Диапазон измерений, показатели неопределенности измерений
Диапазон измерений, мг/дм 3
Суммарная стандартная относительная неопределенность, и, %
Расширенная относительная неопределенность 2 , U при коэффициенте охвата k = 2, %
1 В соответствии с ГОСТ Р 8.563-2009 (п. 3.4) в качестве показателя точности измерений использованы показатели неопределенности измерений).
8.2.3 При отборе проб составляют сопроводительный документ, в котором указывают:
цель анализа, предполагаемые загрязнители;
должность, фамилия отбирающего пробу, дата.
8.3 Подготовка оксида алюминия II степени активности
Перед употреблением оксид алюминия прокаливают в муфельной печи при 600 °С в течение 4 ч. К остывшему оксиду алюминия добавляют к дистиллированную воду (3 % масс.) и выдерживают в течение суток при комнатной температуре.
Срок хранения в эксикаторе или в колбе с притертой пробкой 1 месяц.
9.1 Пробу воды объемом 500 — 1000 см 3 помещают в делительную воронку. Анализируемый раствор должен иметь рН > 2. Перед экстракцией в него вводят 2 — 3 г хлорида натрия. Если разделение слоев после экстракции проходит плохо, количество прибавляемого хлорида натрия увеличивают. Экстрагентом служит петролейный эфир. Экстракцию проводят 2 — 3 раза (время проведения экстракции 1 — 2 мин) порциями по 20 — 25 см 3 органического растворителя, обмывая им стенки всех применяемых стеклянных сосудов: делительной воронки, склянки, в которой была проба.
Экстракты объединяют и сушат прокаленным сернокислым натрием не менее 30 минут 3 .
3 Иногда при использовании петролейного эфира экстракт получается мутным. В этом случае экстракт переносят в мерную колбу, пропуская через смоченный петролейным эфиром фильтр, и промывают остаток на фильтре тем же растворителем.
9.2 Затем экстракт переносят в мерную колбу вместимостью 100 см 3 , сернокислый натрий промывают тремя порциями по 3 см 3 петролейного эфира, которые присоединяются к экстракту. Доводят объем раствора до метки в мерной колбе петролейным эфиром и перемешивают. Отобрав аликвотную порцию экстракта, переносят ее в предварительно взвешенный бюкс и осторожно удаляют органический растворитель нагреванием на водяной бане, в конце при 100 — 105 °С в сушильном шкафу, после чего взвешивают бюкс.
Высушивание и взвешивание продолжают до получения постоянной массы.
По п. 10.1 находят массовую концентрацию всех экстрагированных веществ.
9.3 Для определения массовой концентрации нефтепродуктов другую аликвотную порцию экстракта пропускают через колонку с оксидом алюминия, далее поступают как описано в п. 9.2.
По п. 10.2 находят массовую концентрацию нефтепродуктов.
9.4 При анализе сильно загрязненных сточных вод рекомендуется следующая предварительная обработка пробы. 500 см 3 пробы воды (рН > 2) помещают в фарфоровую или стеклянную чашку, прибавляют 30 г предварительно прокаленного и охлажденного песка, перемешивают и выпаривают на водяной бане досуха. Сухой остаток количественно переносят на бумажный фильтр «синяя лента» или в экстракционный патрон, помещают в аппарат Сокслета, заливают петролейным эфиром и проводят экстракцию в течение 3 — 4 часов. Далее переносят экстракт в мерную колбу и продолжают анализ по п. 9.2.
10.1 Массовую концентрацию всех экстрагированных веществ, X1 (мг/дм 3 ) рассчитывают по формуле:
где m1 — масса бюкса с остатком после удаления петролейного эфира, мг;
m2 — масса пустого бюкса, мг;
V1 — объем аликвотной порции экстракта, см 3 ;
V2 — вместимость мерной колбы с экстрактом, см 3 ;
V — объем пробы, взятой для анализа, см 3 .
10.2 Массовую концентрацию нефтепродуктов Х2 (мг/дм 3 ) рассчитывают по формуле:
где m1 — масса бюкса с остатком после удаления петролейного эфира, мг;
m2 — масса пустого бюкса, мг;
V1 — объем аликвотной порции экстракта, см 3 ;
V2 — вместимость мерной колбы с экстрактом, см 3 ;
V — объем пробы, взятой для анализа, см 3 .
10.3 Массовую концентрацию жиров, X (мг/дм 3 ) находят по разности содержаний экстрагированных веществ и нефтепродуктов (X = X1 — Х2).
10.4 При необходимости за результат измерений Хср принимают среднее арифметическое значение двух параллельных определений Х1 и Х2
для которых выполняется следующее условие:
где r — предел повторяемости, значения которого приведены в таблице 2.
Таблица 2 — Значения предела повторяемости при вероятности Р = 0,95
Диапазон измерений, мг/дм 3
Предел повторяемости (относительное значение допускаемого расхождения между двумя результатами параллельных определений), r, %
При невыполнении условия (4) могут быть использованы методы проверки приемлемости результатов параллельных определений и установления окончательного результата согласно раздела 5 ГОСТ Р ИСО 5725-6-2002.
U — значение показателя точности измерений (расширенная неопределенность измерений с коэффициентом охвата 2).
Значение U приведено в таблице 1.
Допускается результат измерений в документах, выдаваемых лабораторией, представлять в виде: Х ± ,01 ∙ Uл ∙ X, мг/дм 3 , Р = 0,95, при условии Uл 3 .
Процедуру измерений признают удовлетворительной, при выполнении условия:
При невыполнении условия (7) контрольную процедуру повторяют. При повторном невыполнении условия (7) выясняют причины, приводящие к неудовлетворительным результатам, и принимают меры по их устранению.
Расхождение между результатами измерений, полученными в двух лабораториях, не должно превышать предела воспроизводимости. При выполнении этого условия приемлемы оба результата измерений, и в качестве окончательного может быть использовано их среднее арифметическое значение. Значения предела воспроизводимости приведены в таблице 3.
Таблица 3 — Значения предела воспроизводимости при вероятности Р = 0,95
Диапазон измерений, мг/дм 3
Предел воспроизводимости (относительное значение допускаемого расхождения между двумя результатами, полученными в разных лабораториях), R, %
При превышении предела воспроизводимости могут быть использованы методы оценки приемлемости результатов измерений согласно раздела 5 ГОСТ Р ИСО 5725-6-2002.
источник
Аналитической химией называется наука, занимающаяся изучением методов и приемов определения состава веществ и их смесей. Аналитическая химия в целом относится к прикладным наукам, т. е. к наукам, имеющим прикладное практическое значение. Практическое значение аналитической химии весьма разнообразно.
Определение количественного состава исследуемого вещества, т. е. содержания отдельных составных частей его, является задачей количественного анализа. Гравиметрический анализ является одним из методов количественного анализа.
В гравиметрии определяемое вещество осаждают в виде малорастворимого соединения определенной стехиометрии. После выделения и высушивания осадок взвешивают на аналитических весах и по его массе и известной стехиометрии находят количество определяемого компонента.
Гравиметрические методы чрезвычайно точны, потому что на аналитических весах можно взвесить вещества с высокой степенью точности. Массу можно определить до пятой цифры после запятой.
Сущность гравиметрического анализа
Гравиметрическим анализом называют метод количественного химического анализа, основанный на точном измерении массы определяемого вещества или его составных частей, выделяемых в виде соединений точно известного постоянного состава. Гравиметрические определения можно разделить на три группы: методы осаждения, отгонки и выделения.
Методы осаждения основаны на осаждении определяемого компонента в виде малорастворимого химического соединения, фильтровании, прокаливании до постоянной массы и последующем определении массы полученного вещества. При этом различают осаждаемую форму – форму, в виде которой определяемое вещество осаждают, и гравиметрическую форму – форму, в виде которой определяемое вещество взвешивают.
Методы отгонки основаны на отгонке определяемого компонента в виде летучего соединения с последующим определением массы отогнанного вещества (прямое определение) или массы остатка (косвенное определение).
Методы выделения основаны на количественном выделении определяемого компонента из анализируемого раствора путем химической реакции с последующим определением массы выделенного вещества. Этот принцип положен в основу электрогравиметрического метода анализа, в котором определяемый компонент выделяется из раствора в результате электрохимических реакций, протекающих на электродах.
Среди гравиметрических методов анализа наиболее широко применяют метод осаждения.
Методами количественного анализа проверяют правильность технологических процессов, решают многие вопросы исследовательско-прикладного характера: оценивают содержание ценных веществ в рудах, биологических объектах, присутствие токсических веществ в продуктах питания, медицинских препаратах, окружающей среде и т. д.
Весовой анализ основан на том, что из определенного взвешенного количества вещества (навески) посредством соответствующих химических реакций выделяют определенную составную часть в виде нерастворимого осадка. Этот осадок отфильтровывают, промывают и после прокаливания или высушивания взвешивают на аналитических весах. Затем по массе осадка вычисляют количество этой составной части.
Весовой анализ включает несколько этапов:
1. Отбор средней пробы и подготовка вещества к анализу.
5. Определение полноты осаждения (проба на полноту осаждения).
6. Фильтрование и промывание осадка.
7. Определение полноты промывания.
8. Высушивание или прокаливание осадка.
9. Вычисление результатов анализа.
2.2 Механизм реакции осаждения
В процессе образования осадка различают три основных параллельно протекающих процесса:
1) образование зародышей кристаллов;
3) объединение хаотично ориентированных мелких кристаллов.
В начальный момент смешивания реагирующих компонентов раствор, содержащий эти компоненты, пересыщается и образуются мельчайшие частицы осадка – зародыши. Зародыш кристалла – наименьший агрегат атомов, молекул или ионов, который образуется в виде твердой фазы при осаждении и способен к самопроизвольному росту. Образование зародышей в пересыщенном растворе может происходить как самопроизвольно, так и при введении в раствор твердых частиц осадка, которые могут служить центром образования зародышей. Нерастворимые частицы, содержащиеся в реактивах и растворителе, также являются центром образования зародышей. Время с момента смешивания растворов реагирующих веществ до появления зародышей называют индукционным периодом, продолжительность его зависит от концентрации реагирующих веществ, а также от природы осадка. Так, при осаждении творожистого осадка AgCl индукционный период незначителен, а при осаждении кристаллических осадков – достаточно велик.
Рост кристаллов происходит за счет диффузии ионов к поверхности растущего кристалла и осаждения этих ионов на его поверхности и определяется не только диффузионными процессами, но и структурой растущих кристаллов, дефектами кристаллической решетки, внедрением в нее различных ионов и т. д.
Число и размер частиц осадка зависят от соотношения скорости образования зародышей кристаллов и скорости роста кристаллов. Если скорость образования зародышей кристаллов мала по сравнению со скоростью роста кристаллов, образуется небольшое число крупных частиц – осадок крупнокристаллический, при обратном соотношении скоростей получается мелкодисперсный осадок, состоящий из большого числа мелких частиц. Скорости обоих процессов зависят от относительного пересыщения раствора, которое определяется выражением:
где C – концентрация осаждаемого вещества в растворе, получаемая в момент внесения осадителя; S – растворимость.
2.3 Осаждаемая и гравиметрическая формы
При осаждении форма осадка может быть различной в зависимости от условий, в которых оно проводится. Важно подобрать такие условия, при которых не происходит потери вещества. Поэтому осаждение считают важнейшей операцией гравиметрического анализа. При его выполнении необходимо правильно выбрать осадитель, рассчитать его количество, соблюдать определенные условия осаждения, убедиться в полноте осаждения иона из раствора.
Осадок в процессе анализа приходится доводить до постоянной массы. Поэтому в гравиметрическом анализе различают две формы: осаждаемую и гравиметрическую.
Осаждаемая форма – тот осадок, который получается в результате химической реакции между осаждаемым ионом и осадителем.
Например: Ba2+ + SO4 2– → BaSO4
К осаждаемой форме предъявляются следующие требования:
· малая величина растворимости, около 1•10–6 моль/л,
· осадок должен быть крупнокристаллическим,
· осаждаемая форма должна легко и полно превращаться в гравиметрическую форму.
Гравиметрическая форма – то вещество, которое получается после прокаливания осаждаемой формы.
В некоторых случаях осаждаемая и гравиметрическая формы одинаковы (например, BaSO4 ). В других случаях их состав отличается друг от друга:
| Осаждаемая форма | Гравиметрическая форма |
| CaCO3 | CaO |
| Fe(OH)3 | Fe2O3 |
| Al(OH)3 | Al2O3 |
Требования, предъявляемые к гравиметрической форме:
1. Состав гравиметрической формы должен точно соответствовать определенной стехиометрической формуле.
2. Она не должна менять своей массы на воздухе из-за поглощения паров H2 O и CO2 или частичного разложения.
3. Содержание определяемого элемента в гравиметрической форме должно быть как можно меньше, т. к. в таком случае погрешности взвешивания в меньшей степени сказываются на результате.
Перечисленные требования к осадкам в свою очередь определяют требования к осадителям:
1. Осадитель должен образовывать с исследуемым компонентом осадок, обладающий наименьшей растворимостью.
2. Осадитель должен быть летуч, чтобы примеси его можно было удалить при прокаливании.
3. Осадитель должен быть специфичным, т. е. осаждать избирательно.
Влияние ионной силы раствора. В аналитической практике образование и растворение осадка всегда происходит в присутствии посторонних электролитов. Так, при взаимодействии, например, растворов, содержащих стехиометрические количества BaCl2 и Na2SO4, в системе наряду с образовавшимся BaSO4 и одноименными с осадком ионами Ba2+ и SO4 2– будут находиться разноименные с осадком ионы Na+ и Cl– . Нахождение в растворе электролита, содержащего разноименные с осадком ионы, увеличивает ионную силу раствора I. При этом существенное влияние оказывают как концентрация ионов, находящихся в растворе, так и их заряд:
где Ci – молярная концентрация i -го иона; Z – заряд i -го иона.
Таким образом, введение в насыщенный раствор малорастворимого вещества раствора электролита, не содержащего одноименных с малорастворимым веществом ионов, вызывает увеличение растворимости малорастворимого вещества.
Влияние одноименных ионов. Введение в раствор одноименных с осадком ионов приводит к сдвигу равновесия и, соответственно, к уменьшению растворимости осадка.
Следует отметить, что в некоторых случаях при введении в раствор избыточного количества ионов, одноименных с осадком, растворимость осадка может увеличиваться вследствие образования растворимых комплексов.
Влияние pH среды. Если осадок представляет собой соль слабой кислоты, то при добавлении более сильной кислоты анионы осадка, находящиеся в растворе, будут взаимодействовать с ионами водорода с образованием слабой кислоты. При этом равновесие сдвигается вправо за счет протекания реакций и растворимость осадка увеличивается.
Влияние комплексообразующих реагентов. При введении в систему раствор – осадок соединений, образующих устойчивые комплексы с катионами малорастворимого электролита, растворимость осадка увеличивается.
Следует отметить, что на растворимость осадков помимо перечисленных выше факторов также оказывают влияние: 1) температура; 2) применяемый растворитель; 3) конкурирующие окислительно-восстановительные реакции.
Таким образом, для удовлетворения основного требования, предъявляемого к осадку в гравиметрическом анализе, – его малой растворимости – необходимо вести осаждение в присутствии одноименных ионов, при строго определенном pH среды, в отсутствие мешающих комплексообразующих реагентов, окислителей или восстановителей, необходимо контролировать температуру, при которой проводится осаждение.
Основной причиной загрязнения осадка является соосаждение. Соосаждением называют одновременное осаждение растворимого компонента с макрокомпонентом из одного и того же раствора путем адсорбции, окклюзии, образования смешанных кристаллов или механического захвата частиц других фаз. Осадки при этом загрязнены веществами, произведение растворимости для которых не достигается.
Адсорбция – увеличение поверхностной концентрации растворенных веществ на границе раздела фаз. В соответствии с правилом адсорбции на поверхности осадка в первую очередь адсорбируются ионы, входящие в состав кристаллической решетки осадка и находящиеся в избытке. Под действием заряда к поверхности осадка притягиваются противоионы, которые удерживаются слабее первично адсорбированных ионов.
Адсорбируемость ионов на поверхности осадка зависит также от концентрации ионов, находящихся в растворе, от заряда ионов и от их размера. Количество адсорбированных на поверхности осадка ионов тем больше, чем больше его поверхность, поэтому к адсорбции более склонны осадки с развитой поверхностью, т. е. аморфные. Для предотвращения явления адсорбции осаждение как аморфных, так и кристаллических осадков проводят в условиях, позволяющих получить осадки с наименьшей поверхностью; повышение температуры также способствует уменьшению адсорбции, так как адсорбция – экзотермический процесс. Количество адсорбированных примесей можно уменьшить при промывании осадков на фильтре водой или промывной жидкостью, а также в случае кристаллических осадков в процессе их старения.
Окклюзия – процесс включения посторонних веществ внутрь осадков в ходе их образования. Окклюзия характерна для кристаллических осадков и наблюдается при быстром росте кристаллов, когда часть противоионов, адсорбированных на поверхности растущего кристалла, остается внутри его. Окклюдированные примеси не удаляются промыванием, но окклюзию можно уменьшить путем переосаждения осадка, а также в процессе его старения. Степень окклюзии в процессе осаждения можно уменьшить медленным добавлением осадителя по каплям, при перемешивании.
Смешанные кристаллы – кристаллы, содержащие второй компонент, внедряющийся в решетку основного кристалла и распределенный в этой решетке. Механический захват – процесс случайного включения относительно малых количеств других фаз внутрь осадка в ходе его образования. Механический захват обусловлен несовершенством кристаллической решетки осадка, наличием в ней пустот и трещин при быстром росте кристаллов. Для уменьшения механического захвата необходимо осаждать кристаллические осадки из разбавленных растворов, добавляя осадитель медленно по каплям, при перемешивании. Переосаждение, а также старение кристаллических осадков тоже способствует устранению механического захвата примесей.
Причиной загрязнения осадков может служить также последующее осаждение, в ходе которого на поверхности ранее выделенного осадка осаждается химически отличающаяся от него форма соединения, содержащего ион, одноименный с осадком.
2.6 Получение осаждаемой формы
Условия образования кристаллических осадков
| Условия осаждения | Достигаемый эффект |
| 1. Осаждение ведут из достаточно разбавленного исследуемого раствора разбавленным раствором осадителя. | 1. Выпадение осадка замедляется, что способствует образованию крупных кристаллов, уменьшается осаждение. |
| 2. Раствор осадителя прибавляют медленно, по каплям, при постоянном помешивании стеклянной палочкой. | 2. Капли раствора осадителя разбавляются большим объемом анализируемого раствора, вследствие чего предотвращаются местные пересыщения, осадок увлекает меньше примесей осадителя. |
| 3. Осаждение ведут из подогретого исследуемого раствора горячим раствором осадителя. | 3. Повышение температуры в процессе осаждения ускоряет формирование кристаллической решетки и тормозит образование зародышевых центров кристаллизации. |
| 4. Прибавлять при осаждении вещества, которые повышают растворимость осадка. | 4. Повышается растворимость образующегося соединения, меньше образуется первичных кристаллов и тем крупнее они будут. |
| 5. Отфильтровать осадок после охлаждения раствора. | 5. Снижается растворимость; имеет место более полное осаждение. |
| 6. После прибавления осадителя оставить осадок на несколько часов. | 6. Происходит созревание осадка – растворение мелких кристаллов и рост крупных. При этом удаляются первоначально включенные в осадок примеси. Устраняются дефекты кристаллической решетки. |
В таблицах приведены условия осаждения кристаллических и аморфных осадков, применяемых в гравиметрическом анализе. Осадки, получаемые в этих условиях, удовлетворяют основным требованиям, предъявляемым к ним.
Условия образования аморфных осадков
| Условия осаждения | Достигаемый эффект |
| 1. Осаждение ведут в присутствии электролита – коагулятора. | 1. Добавление в раствор электролита приводит к коагуляции осаждаемого вещества вследствие адсорбции ионов электролита на поверхности частиц. |
| 2. Осаждение ведут из нагретого анализируемого раствора нагретым раствором осадителя. | 2. Повышение температуры способствует разрушению гидратных оболочек коллоидных частиц и десорбции ионов, придающих одноименный заряд коллоидным частицам. |
| 3. Осаждение ведут из достаточно концентрированного исследуемого раствора концентрированным раствором осадителя. | 3. Из-за небольшого объема раствора получается не слишком объемистый осадой; уменьшается адсорбция осадком примесей из раствора; разрушаются гидратные оболочки коллоидных частиц. |
2.7 Фильтрование и промывание осадка
Выбор приспособлений для фильтрования зависит от природы осадка и от температуры, при которой осадок переводят в гравиметрическую форму. Во всех случаях фильтрования осадка сопутствует его промывание. Промывание необходимо для удаления ионов, которые не улетучиваются при переводе осадка в гравиметрическую форму.
При промывании аморфных осадков дистиллированной водой происходит их пептизация, т. е. переход в коллоидное состояние, коллоидные частицы проходят через поры фильтра в промывные воды, отфильтровать осадок не удается. Поэтому промывная жидкость для аморфных осадков должна содержать электролиты-коагуляторы, препятствующие пептизации, такие как разбавленные растворы летучих кислот (HNO3 ), растворы солей аммония (NH4 Cl, NH4 NO3 и др.). Кроме того, адсорбированные на поверхности осадка и загрязняющие его ионы при промывании осадка указанными промывными жидкостями замещаются ионами, способными улетучиваться при прокаливании.
Промывная жидкость для кристаллических осадков обычно содержит летучие электролиты; осадки веществ с растворимостью 10–5 –10–6 моль/л промывают растворами электролитов, содержащих одноименные с осадком ионы.
Общий объем промывной жидкости не должен превышать 100 мл. Осадок более полно освобождается от загрязняющих веществ, если его промывать многократно небольшими порциями промывной жидкости, чем при двух-трехкратном промываниями большими порциях, что можно видеть из следующего расчета:
где Cn – концентрация примесей после n -го промывания; C – начальная концентрация примесей; V – объем промывной жидкости, не стекающей через фильтр; V – объем каждой порции промывной жидкости.
Осадок сначала промывают в стакане, в котором проводили осаждение, методом декантации. Затем осадок переносят на фильтр и промывают на нем небольшими порциями промывной жидкости.
2.8 Получение гравиметрической формы
Гравиметрическая форма может быть получена путем высушивания осадка или прокаливанием его до постоянной массы. Высушивание осадка проводят при применении органических осадителей, при этом гравиметрическая форма совпадает с формой осаждения. При прокаливании гравиметрическая форма может взаимодействовать с углеродом с изменением формулы соединения. Так, при прокаливании BaSO4 возможна следующая реакция:
В этом случае необходимо продолжить прокаливание на воздухе для окисления сульфида бария в сульфат кислородом воздуха.
Температура прокаливания зависит от природы осадка. Для того чтобы выбрать температуру прокаливания, снимают термогравиметрическую кривую. При этом с помощью автоматических термовесов непрерывно фиксируют массу осадка по мере равномерного возрастания температуры в печи. Температура, пригодная для прокаливания, соответствует горизонтальному участку кривой.
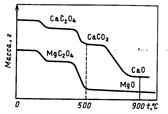 Термогравиметрические кривые для раздельного определения кальция и магния. Термогравиметрические кривые можно применять для раздельного определения компонентов смесей. Различный ход термогравиграмм дает возможность определять содержание компонентов смеси при их совместном осаждении. Путем расчетов можно определить содержание в смеси обоих компонентов.
Термогравиметрические кривые для раздельного определения кальция и магния. Термогравиметрические кривые можно применять для раздельного определения компонентов смесей. Различный ход термогравиграмм дает возможность определять содержание компонентов смеси при их совместном осаждении. Путем расчетов можно определить содержание в смеси обоих компонентов.
2.9 Применение гравиметрического метода анализа
Методы осаждения часто применяют как методы разделения. Гравиметрический анализ по методу осаждения применяют при анализе эталонов для калибровки и контроля физико-химических методов анализа, при определении состава синтезированных соединений и др.
Существует также ряд специфических гравиметрических методов определения органических соединений. Например, при определении содержания фенолфталеина его осаждают из щелочного раствора в виде тетраиодида, высушивают и взвешивают.
При достаточно большой разности в произведениях растворимости двух осадков возможно их последовательное осаждение и разделение (ПР1 :ПР2 ≥104 ). При этом первым начнет осаждаться тот ион, для которого быстрее достигается произведение растворимости. Однако последовательное осаждение не всегда обеспечивает полноту осаждения разделяемых компонентов.
Гравиметрические методы анализа менее избирательны, чем другие методы анализа. Избирательность может быть повышена при использовании органических аналитических реагентов-осадителей, реакций внешнесферного комплексообразования, приемов маскирования, регулирования pH среды и др.
Весовые определения
Схема гравиметрического анализа по методу осаждения предусматривает последовательное выполнение следующих основных операций:
1) отбор пробы и подготовка ее к анализу;
3) переведение навески вещества в раствор;
4) получение осаждаемой формы;
8) получение гравиметрической формы.
Стаканы. В гравиметрическом методе анализа применяют химические стаканы различной вместимости. Для осаждения кристаллических осадков обычно применяют стаканы с носиком вместимостью 200-250 мл, для осаждения аморфных осадков – стаканы вместимостью 100-150 мл. При одинаковой вместимости стаканы могут быть различной высоты, лучше применять более низкие стаканы, т. к. их дно легче очищать от осадка.
Воронки, применяемые для фильтрования, могут быть различного диаметра, в зависимости от количества отделяемого осадка: они должны иметь наклон стенок 60° и удлиненный косо срезанный конец, внутренний диаметр которого в верхней части меньше, чем в нижней, благодаря этому увеличивается скорость фильтрования и промывания осадка.
Тигли. Фарфоровые тигли применяют для высокотемпературного прокаливания осадков. Фарфоровые тигли можно нагревать до температур не выше 1200°C. Кроме фарфоровых тиглей в гравиметрическом анализе для высокотемпературного сплавления и прокаливания металлические, кварцевые и другие тигли. До окончания всех операций тигли нельзя брать руками, а только при помощи металлических щипцов.
Стеклянные фильтрующие тигли представляют собой стеклянные тигли с вплавленными фильтрующими пластинками из прессованного пористого стекла. Их применяют для фильтрования с последующим высушиванием в сушильном шкафу осадков, которые разлагаются при высоких температурах.
Эксикаторы применяют для охлаждения тиглей при доведении их массы до постоянного значения, а также для хранения прокаленных тиглей и высушивания. В качестве осушителя в эксикаторах чаще всего применяют безводный хлорид кальция, реже – концентрированную H2 SO4 , P2 O5 и др. При работе с эксикатором необходимо соблюдать следующие правила: 1) необходимо следить, чтобы притертые части всегда были смазаны; 2) перенося эксикатор, обязательно следует придерживать его крышку; 3) поместив горячий тигель в эксикатор, крышку эксикатора оставляют приоткрытой в течение 3-5 минут, пока воздух внутри эксикатора не прогреется; 4) нельзя оставлять эксикатор открытым; 5) открывая и закрывая эксикатор, крышку следует сдвигать в сторону, а не поднимать.
Кроме перечисленной посуды применяют также стеклянные палочки обычные и с резиновыми насадками, часовые стекла для накрывания стаканов с осадками, промывалки, мерные цилиндры и др.
3.2 Определение кристаллизационной воды в кристаллическом хлориде бария
Кристаллизационной водой называется вода, входящая в структуру кристаллов некоторых веществ, называемых кристаллогидратами. Содержание кристаллизационной воды определяют высушиванием кристаллогидрата до постоянной массы. Температура, при которой происходит удаление кристаллизационной воды, зависит от прочности связи ее с основным веществом. Так, щавелевая кислота H2C2O4 •2H2O сушится при 110-120°C, медный купорос CuSO4•5H2 O – при 140-150°C, алюмокалиевые квасцы KAl(SO4 )•12H2 O около 230°C, хлорид бария BaCl2 •2H2O – при 120-125°C, сода Na2CO3 •10H2O – около 270°C, а глауберова соль Na2 SO4 •10H2 O – при температуре выше 300°C.
Вещество, предназначенное для определения кристаллизационной воды, должно быть воздушно-сухим. Иначе вместе с кристаллизационной водой будет определена и гигроскопическая, т. е. адсорбционная вода.
1. Берут чистый бюкс, маркируют его графитовым карандашом на пришлифованной части и помещают в сушильный шкаф с температурой 120-125°C.
2. Через 45-60 мин. Помещают бюкс с помощью тигельных щипцов в эксикатор. Когда бюкс остынет до температуры аналитических весов, взвешивают его и записывают результат в лабораторный журнал.
3. Повторяют высушивание бюкса еще 1-2 раза по 30 мин., чтобы довести его до постоянной массы. Высушивание заканчивают, когда результаты двух последних взвешиваний будут отличаться не более, чем на 0,0002 г.
4. В подготовленный бюкс помещают 1,5-2,0 г свежеперекристаллизованного воздушно-сухого хлорида бария BaCl2 •H2 O и взвесьте на аналитических весах.
1. Помещают бюкс в сушильный шкаф. Первое высушивание соли проводится 1,5-2,0 часа, строго следя, чтобы температура находилась в пределах 120-125°C. При более высокой температуре возможно частичное разложение и улетучивание соли, а при более низкой – не вся кристаллизационная вода будет удалена.
2. Затем переносят бюкс в эксикатор, оставляют охлаждаться на 15-20 мин., т. е. доводят бюкс с его содержимым до постоянной массы.
Вычисление процентного содержания воды и ошибки анализа
По результатам измерений определяют процентное содержание воды, абсолютную и относительную ошибки анализа:
1.Теоретическое содержание воды:
2. Практическое содержание воды:
a – навеска кристаллогидрата хлорида бария
b – масса безводного хлорида бария.
3. Абсолютная ошибка – это разность между найденным результатом анализа и действительным содержанием: Δ=14,68% – 14,75%= – 0,07%
4. Относительная ошибка – отношение абсолютной ошибки к истинному содержанию воды. Выражается она обычно в процентах и считается положительной величиной:
3.3Определение содержания железа в растворе хлорида железа (III)
Последовательность выполнения работы:
1. В чистый химический стакан берут для анализа раствор FeCl3 .
2. Подкисляют его 3-5 мл 2 н. раствора HNO3 и осторожно нагревают, не допуская кипения.
3. К горячему раствору прибавляют по каплям 10% раствор аммиака до слабого, но ощутимого запаха.
4. Затем содержимое стакана тщательно перемешивают палочкой, разбавляют 100 мл горячей дистиллированной воды и еще раз перемешивают.
5. Дают осадку отстояться, а когда раствор над ним станет совершенно прозрачным, делают пробу на полноту осаждения 1-2 каплями раствора аммиака.
2. Фильтрование и промывание:
1. Убедившись в полноте осаждения приступают к фильтрованию. Для этого используют неплотный фильтр. Декантируют жидкость на фильтр, осадок промывают в стакане 2-3 раза 2% горячим раствором NH4 NO3 .
2. Количественно без потерь переносят осадок на фильтр и продолжают промывать до отрицательной реакции фильтрата с нитратом серебра (в присутствии HNO3 ) на ион Cl– .
3. Высушивание и прокаливание:
1. Фильтр с осадком подсушивают в сушильном шкафу и слегка влажным переносят в тигель, предварительно доведенный до постоянной массы.
2. Осторожно озоляют фильтр на электроплитке, следя, чтобы он не вспыхнул.
3. Затем помещают тигель в муфельную печь и прокаливают до постоянной массы.
Вычисление результатов анализа
Составляют уравнение реакций:
FeCl3 + 3NH4 OH → Fe(OH)3 ↓ + 3NH4 Cl
Предположим, что при анализе получены следующие данные:
масса тигля с осадком (первое взвешивание) — 16,3242 г
масса тигля с осадком (второе взвешивание) — 16,3234 г
масса тигля с осадком (третье взвешивание) — 16,3232 г
159,68 г (Fe2 O3 ) соответствуют 324,24 г (FeCl3 )
0,1702 г (Fe2 O3 ) соответствуют Х г (FeCl3 )
В 162,21 г FeCl3 содержится 55,85 г Fe
В 0,3457 г FeCl3 содержится Х г Fe
Используют величину фактора пересчета:
a – масса прокаленного осадка,
F – фактор пересчета или аналитический множитель.
Гравиметрический анализ – один из наиболее универсальных методов. Он применяется для определения почти любого элемента. Гравиметрические методы чрезвычайно точны, потому что на аналитических весах можно взвесить вещества с высокой степенью точности. Массу можно определить до пятой цифры после запятой.
Гравиметрический анализ – важнейший метод количественного химического анализа, в котором взвешивание является не только начальной, но и конечной стадией определения. Гравиметрический анализ сыграл большую роль при установлении закона постоянства состава химических соединений, закона кратных отношений, периодического закона и др.
Чаще всего гравиметрический метод применяют для определения основных компонентов пробы, когда на выполнение анализа отводится несколько часов или десятков часов, для анализа эталонов, используемых в других методах, в арбитражном анализе, для установления состава минералов.
Список литературы
1. К.М.Ольшанова, С.К. Пискарева, К.М.Барашков «Аналитическая химия», Москва, «Химия», 1980 г.
2. «Аналитическая химия. Химические методы анализа», Москва, «Химия», 1993 г.
3. Аналитическая химия: Учебно-методическое пособие для студентов педагогических вузов.-Самара: Изд-во СамГПУ, 2007
4. Аналитическая химия. Химические методы анализа/Под. ред. О. М. Петрухина. М.: Химия, 1992. 400 с. Ил.
5. Бончев П. Р. Введение в аналитическую химию. Л.: Химия, 1978. 496 с.
Дата добавления: 2017-02-24 ; просмотров: 6755 | Нарушение авторских прав
источник