1. Атомно-абсорбционный анализ
2. Достоинства атомно-абсорбционного анализа
Метод атомно-абсорбционного спектрального анализа отличается высокой абсолютной и относительной чувствительностью. Метод позволяет с большой точностью определять в растворах около восьмидесяти элементов в малых концентрациях, поэтому он широко применяется в биологии, медицине (для анализа органических жидкостей), в геологии, почвоведении (для определения микроэлементов в почвах) и других областях науки, а также в металлургии для исследований и контроля технологических процессов.
По точности и чувствительности этот метод превосходит многие другие; поэтому его применяют при аттестации эталонных сплавов и геологических пород (путем перевода в раствор).
Атомно-абсорбционный анализ (атомно-абсорбц. спектрометрия), метод количественного элементного анализа по атомным спектрам поглощения (абсорбции). Через слой атомных паров пробы, получаемых с помощью атомизатора, пропускают излучение в диапазоне 190-850 нм. В результате поглощения квантов света атомы переходят в возбужденные энергетические состояния. Этим переходам в атомных спектрах соответствуют резонансные линии, характерные для данного элемента. Согласно закону Бугера-Ламберта-Бера, мерой концентрации элемента служит оптическая плотность A = lg(I0/I), где I0 и I-интенсивности излучения от источника соответственно до и после прохождения через поглощающий слой.
Принципиальная схема пламенного атомно-абсорбционного спектрометра: 1-источник излучения; 2-пламя; 3-монохрома гор; 4-фотоумножитель; 5-регистрирующий или показывающий прибор.
Приборы для атомно-абсорбционного анализа — атомно-абсорбционные спектрометры — прецизионные высокоавтоматизированные устройства, обеспечивающие воспроизводимость условий измерений, автоматическое введение проб и регистрацию результатов измерения. В некоторые модели встроены микроЭВМ. В качестве примера на рис. приведена схема одного из спектрометров. Источником линейчатого излучения в спектрометрах чаще всего служат одноэлементные лампы с полым катодом, заполняемые неоном. Для определения некоторых легколетучих элементов (Cd, Zn, Se, Те и др.) удобнее пользоваться высокочастотными безэлектродными лампами.
Перевод анализируемого объекта в атомизированное состояние и формирование поглощающего слоя пара определенной и воспроизводимой формы осуществляется в атомизаторе — обычно в пламени или трубчатой печи. Наиболее часто используют пламя смесей ацетилена с воздухом (макс. т-ра 2000°С) и ацетилена с N2O (2700°С). Горелку со щелевидным соплом длиной 50-100 мм и шириной 0,5-0,8 мм устанавливают вдоль оптич. оси прибора для увеличения длины поглощающего слоя.
Трубчатые печи сопротивления изготавливают чаще всего из плотных сортов графита. Для исключения диффузии паров через стенки и увеличения долговечности графитовые трубки покрывают слоем газонепроницаемого пироуглерода. Максимальная температура нагрева достигает 3000 °С. Менее распространены тонкостенные трубчатые печи из тугоплавких металлов (W, Та, Мо), кварца с нихромовым нагревателем. Для защиты графитовых и металлических печей от обгорания на воздухе их помещают в полугерметичные или герметичные камеры, через которые продувают инертный газ (Аr, N2).
Введение проб в поглощающую зону пламени или печи осуществляют разными приемами. Растворы распыляют (обычно в пламя) с помощью пневматических распылителей, реже — ультразвуковых. Первые проще и стабильнее в работе, хотя уступают последним в степени дисперсности образующегося аэрозоля. Лишь 5-15% наиболее мелких капель аэрозоля поступает в пламя, а остальная часть отсеивается в смесительной камере и выводится в сток. Максимальная концентрация твердого вещества в растворе обычно не превышает 1%. В противном случае происходит интенсивное отложение солей в сопле горелки.
Термическое испарение сухих остатков растворов — основной способ введения проб в трубчатые печи. При этом чаще всего пробы испаряют с внутренней поверхности печи; р-р пробы (объемом 5-50 мкл) вводят с помощью микропипетки через дозировочное отверстие в стенке трубки и высушивают при 100°С. Однако пробы испаряются со стенок при непрерывном возрастании температуры поглощающего слоя, что обусловливает нестабильность результатов. Чтобы обеспечить постоянство температуры печи в момент испарения, пробу вводят в предварительно нагретую печь, используя угольный электрод (графитовую кювету) графитовый тигель (печь Вудриффа), металлический или графитовый зонд. Пробу можно испарять с платформы (графитового корытца), которую устанавливают в центре печи под дозировочным отверстием. В результате значительного отставания температуры платформы от температуры печи, нагреваемой со скоростью ок. 2000 К/с, испарение происходит при достижении печью практически постоянной температуры.
Для введения в пламя твердых веществ или сухих остатков растворов используют стержни, нити, лодочки, тигли из графита или тугоплавких металлов, помещаемые ниже оптич. оси прибора, так что пары пробы поступают в поглощающую зону с потоком газов пламени. Графитовые испарители в ряде случаев дополнительно подогревают электрическим током. Для исключения мех. потерь порошкообразных проб в процессе нагрева применяются испарители типа цилиндрических капсул, изготовленные из пористых сортов графита.
Иногда растворы проб подвергают в реакционном сосуде обработке в присут. восстановителей, чаще всего NaBH4. При этом Hg, напр., отгоняется в элементном виде, As, Sb, Bi и др.-в виде гидридов, к-рые вносятся в атомизатор потоком инертного газа. Для монохроматизации излучения используют призмы или дифракционные решетки; при этом достигают разрешения от 0,04 до 0,4 нм.
При атомно-абсорбционном анализе необходимо исключить наложение излучения атомизатора на излучение источника света, учесть возможное изменение яркости последнего, спектральные помехи в атомизаторе, вызванные частичным рассеянием и поглощением света твердыми частицами и молекулами посторонних компонентов пробы. Для этого пользуются различными приемами, например модулируют излучение источника с частотой, на которую настраивают приемно — регистрирующее устройство, применяют двухлучевую схему или оптич. схему с двумя источниками света (с дискретным и непрерывным спектрами). наиб. эффективна схема, основанная на зеемановском расщеплении и поляризации спектральных линий в атомизаторе. В этом случае через поглощающий слой пропускают свет, поляризованный перпендикулярно магнитному полю, что позволяет учесть неселективные спектральные помехи, достигающие значений А = 2, при измерении сигналов, которые в сотни раз слабее.
1. Достоинства атомно-абсорбционного анализа
Достоинства атомно-абсорбционного анализа — простота, высокая селективность и малое влияние состава пробы на результаты анализа. Ограничения метода — невозможность одновременного определения нескольких элементов при использовании линейчатых источников излучения и, как правило, необходимость переведения проб в р-р.
Атомно-абсорбционный анализ применяют для определения около 70 элементов (гл. обр. металлов). Не определяют газы и некоторые др. неметаллы, резонансные линии которых лежат в вакуумной области спектра (длина волны меньше 190 нм). С применением графитовой печи невозможно определять Hf, Nb, Та, W и Zr, образующие с углеродом труднолетучие карбиды. Пределы обнаружения большинства элементов в р-рах при атомизации в пламени 1-100мкг/л, в графитовой печи в 100-1000 раз ниже. Абсолютные пределы обнаружения в последнем случае составляют 0,1-100 пг. Относительно стандартное отклонение в оптимальных условиях измерений достигает 0,2-0,5% для пламени и 0,5-1,0% для печи. В автоматическом режиме работы пламенный спектрометр позволяет анализировать до 500 проб в час, а спектрометр с графитовой печью-до 30 проб. Оба варианта часто используют в сочетании с предварит. разделением и концентрированием экстракцией, дистилляцией, ионным обменом, хроматографией, что в ряде случаев позволяет косвенно определять некоторые неметаллы и орг. соединения.
Методы атомно-абсорбционного анализа применяют также для измерения некоторых физических и физ.-химических величин — коэффициент диффузии атомов в газах, температур газовой среды, теплот испарения элементов и др.; для изучения спектров молекул, исследования процессов, связанных с испарением и диссоциацией соединений.
1. Львов Б. В., Атомно-абсорбционный спектральный анализ, М, 1966;
2. Прайс В., Аналитическая атомно-абсорбционная спектроскопия, пер. с англ., М., 1976;
3. Харламов И.П., Еремина Г. В., Атомно-абсорбционный анализ в черной металлургии, М., 1982;
4. Николаев Г. И., Немец А. М., Атомно-абсорбционная спектроскопия в исследовании испарения металлов, М., 1982;
5. Хавезов И., Цалев Д., Атомно-абсорбционный анализ, пер. с болг., Л., 1983. Б. В. Львов. Л. К. Ползик.
источник
Атомно-абсорбционный анализ основан на поглощении резонансного излучения элемента его невозбужденными атомами, находящимися в свободном состоянии, т.е. в состоянии «атомного пара». В результате поглощения квантов света валентные электроны атома возбуждаются и переходят из основного состояния на ближайшее возбужденное, а резонансное излучение с интенсивностью I, проходящее через слой «атомного пара», ослабляется в соответствии с основным законом светопоглощения (законом Бугера-Ламберта-Бера):
, (2.4)
где I – интенсивность резонансного излучения, прошедшего через поглощающий слой плазмы; kv – коэффициент поглощения излучения; l – толщина (длина) поглощающего слоя плазмы; с – концентрация поглощающих атомов.
После логарифмирования уравнения (2.4) и перехода к десятичным логарифмам получаем:
,
где . Величину lg(I/I) называют оптической плотностью и обозначают А, которая и является аналитическим сигналом.
Линейная зависимость A=k´cl лежит в основе всех количественных определений в ААСА. Приведенные выше уравнения справедливы только для монохроматического излучения и в отсутствии химических и физических помех.
Наиболее важным звеном в ААСА является атомизация анализируемой пробы, т.е. перевод вещества в атомарное состояние. Для этой цели применяют, в основном, два типа атомизаторов – пламя и электротермические импульсные атомизаторы.
Пламя. Пламенный атомизатор представляет собой горелку со щелевидной насадкой, выполняющей роль кюветы для атомизируемой пробы. Такие горелки обеспечивают стабильное и безопасное пламя, имеющее температуру от 800 до 3000 °С, достаточную для атомизации большинства элементов. Они обеспечивают хорошую воспроизводимость (sr=0.005–0.05) и низкие пределы обнаружения (сmin=10 –6 –10 –7 % масс.) элементов. Пламенная атомизация с использованием низкотемпературных пламен значительно уменьшает возможность появления поглощаемого резонансного излучения самого атомизируемого вещества, обеспечивает достаточно высокую скорость анализа при небольшой трудоемкости работы.
Электротермические атомизаторы. В настоящее время разработано несколько типов электротермических атомизаторов, действующих на принципе импульсного термического испарения вещества (графитовая печь, кювета Львова, лазерный испаритель и др.), из которых наиболее удачным оказалась графитовая кювета, предложенная Б.В. Львовым в конце 50-х годов прошлого века. Это устройство представляет собой небольшую графитовую трубку, помещенную в инертную атмосферу, которая нагревается током большой силы при низком напряжении, имеет изотермическую зону, где и происходит атомизация. Основное преимущество графитовой кюветы – значительное повышение чувствительности определений вследствие увеличения эффективности атомизации и очень малого объема вводимой пробу. Пределы обнаружения элементов с использованием графитовой кюветы достигают 10 –11 –10 –13 г. В сравнении с пламенными атомизаторами электротермические уступают им в воспроизводимости полученных результатов, сложней в обращении и требуют больших затрат электроэнергии.
Источники излучения. В ААСА применяется источник внешнего излучения. Главное требование к нему – стабильность и высокая степень монохроматизации излучения. В качестве источников резонансного излучения используют газоразрядные лампы низкого давления, лазеры с перестраиваемой частотой, но чаще всего – лампы с полым катодом, испускающие излучение, состоящее из дискретных линий металла катода и газа наполнителя. Лампы с полым катодом представляют собой стеклянный или кварцевый баллон, заполненный инертным газом низкого давления. Внутри расположены два электрода – катод и анод. Катод представляет собой чашу, как правило, из того металла или сплава, элемент которого необходимо определять. При подаче напряжения на электроды возникает тлеющий разряд с образованием положительных ионов газа-наполнителя. Эти ионы бомбардируют катод, выбивая атомы элемента в газовую фазу, где они возбуждаются и дают дискретное излучение, характерное для свободных атомов определяемого элемента. В основном используют одноэлементные лампы, хотя находят применение и многоэлементные лампы с различными раздельнопитаемыми кольцеобразными катодами.
Измерение поглощения излучения. Принципиальная блок-схема измерения поглощения излучения атомами определяемого элемента показана на рис. 2.12. Резонансное линейчатое излучение с интенсивностью I от источника 3 проходит через поглощающий слой 4 атомного пара и монохроматор 5. Ослабленное излучение с интенсивностью I направляется на фотоэлемент (или фотоумножитель) 6, в котором излучение преобразуется в фототок и после его усиления электронным усилителем 7 измеряется регистрирующим устройством 8, измерительная шкала которого обычно проградуирована в значениях оптической плотности.
Рисунок 2.12 – Принципиальная блок-схема атомно-абсорбционного прибора: 1 – анализируемый раствор; 2 – распылитель; 3 – лампа с полым катодом; 4 – пламя горелки; 5 – монохроматор; 6 – фотоэлемент; 7 – усилитель фототока; 8 – регистрирующее устройство
Аппаратное оформление этой схемы может осуществляться в двух вариантах.
1. В однолучевых приборах, как показано на схеме, поглощение излучения атомами определяемого элемента и холостым фоновым «паром», не содержащим поглощающих атомов этого элемента, измеряется поочередно (раздельно) и разность в поглощении учитывается регистрирующим устройством или самим экспериментатором.
2. В двухлучевых приборах резонансное излучение от источника света с помощью зеркал распределяется на два параллельных потока, один из которых проходит через поглощающий слой атомов анализируемого вещества, а второй проходит через фоновый «пар». Регистрирующее устройство поглощение фоном учитывает автоматически или путем соответствующей компенсации.
Определение концентрации анализируемого раствора
Для определения концентрации анализируемых растворов применяют общие для ФХМА методы (способы): метод сравнения, метод градуировочного графика и метод добавок. В большинстве случаев предпочтение отдается методу градуировочного графика А=f(c), построенному по результатам измерения оптических плотностей стандартных растворов. Метод добавок применяют в тех случаях, когда состав пробы неизвестен и наблюдается заметное влияние матричного эффекта.
Важным достоинством метода ААСА является высокая избирательность, поскольку число резонансных линий в спектре невелико и практически отсутствует наложение аналитических линий. Метод обладает экспрессностью, высокой чувствительностью и хорошей воспроизводимостью результатов. Погрешность определения составляет 1–4%. Диапазон определяемых концентраций, как правило, не превышает двух порядков, т.к. он лимитируется величиной оптической плотности, где соблюдается линейная зависимость А=f(c).
К недостаткам метода ААСА следует отнести сложность осуществления многоэлементного анализа, поскольку для каждого элемента нужен свой источник резонансного излучения. Метод ААСА применяется, в основном, для анализа растворов. При использовании электротермических атомизаторов можно анализировать и мелкодисперсные порошкообразные материалы.
По технике выполнения ААСА близок к эмиссионной фотометрии пламени, но обладает более широкими возможностями. Этим методом определяют около 70 элементов с достаточно высокой чувствительностью.
Не нашли то, что искали? Воспользуйтесь поиском:
источник
АТОМНО — АБСОРБЦИОННЫЙ МЕТОД АНАЛИЗА ВОДЫ
Метод атомно — абсорбционной спектроскопии, обладающий высокой экспресностью и хорошей точностью, с успехом применяют для анализа природных и сточных вод. Преимущество метода атомной абсорбции перед многими методами анализа вод состоит в его высокой селективности, низких пределах обнаружения элементов, в простоте подготовки проб к анализу, поскольку в большинстве случаев отпадает необходимость проведения операций, связанных с отделением мешающих элементов, а также в универсальности конечной продукции анализа, т. е, возможности определения нескольких элементов — примесей из одного раствора по единой методике с получением конечных результатов в единицах концентрации. Это обеспечило ему широкое применение в самых различных областях науки, промышленности, сельского хозяйства. И тем не менее метод атомно – абсорбционного анализа при всех его достоинствах не следует считать универсальным, способным заменить все остальные, paнее известные методы анализа. Так, при анализе больших партий однотипных проб, когда возможно прямое определение какого-либо химического элемента, метод пламенной атомно-абсорбционной спектроскопии по производительности обычно превосходит титрометрические и спектрофотометрические методы. Он может быть также успешно применен при анализе проб нестандартного состава в случае относительно больших концентраций определяемых элементов. Однако, этому методуприсущи некоторые недостатки. При необходимости одновременного определения нескольких элементов для каждого из них требуется отдельный просвечивающий источник света, т. е. предполагается последовательное определение отдельных элементов. Как и в методе атомно-эмиссионной спектроскопии, для получения надежных результатов анализа следует учитывать эффект матрицы и стремиться к соответствию эталонов и проб.
ФИЗИЧЕСКИЕ ОСНОВЫ МЕТОДА
Метод атомно-абсорбционной спектроскопии основан на измерении поглощения резонансной линии свободными атомами
определяемого элемента при прохождении света через атомный
пар исследуемого образца. Свободные атомы элементов, находящиеся _в_ невозбужденном стабильном состоянии, в слое нагретого газа-плазмы способны селективно поглощать световую энергию, переходя при этом из нижнего Rв верхнее (возбужденное) состояние с энергией Ei. (Частоты линий поглощения определяются правилом Бора:
В результате облучения атомного пара светом появляется сравнительно небольшое число линий поглощения. Их называют резонансными и используют в качестве аналитических. Таким образом, высокая селективность метода основана на способности свободных атомов поглощать только свойственную данному элементу световую энергию.
Поглощение описывается уравнением Бугера — Ламберта — Бера:
— I oи I v— интенсивности излучения падающего на атомный пар и поглощенного света, т. е. прошедшего через поглощающий спой атомного пара, длина которого равна /;
— с-концентрация атомов поглощающего элемента в атом-iiov паре;
-kv— коэффициент абсорбции, характеризующий чувствительность аналитической линии с частотой v при данных условиях измерения.
Измерив и рассчитав (/v//o) 100%, называемую пропусканием Т, можно рассчитать концентрацию элемента с в поглощающем слое. Однако для простоты расчетов чаще пользуются величиной, называемой оптической плотностью и представляющей собой логарифм величины, обратной Т, т. е. lg I / Т = lg Io / Iv. В результате преобразования уравнение Бугера -Ламберта -Бера принимает вид:
Поскольку длина поглощающего слоя l — величина постоянная ,для данного атомно-абсорбционного спектрометра, а содержание определяемого элемента в пробе пропорционально его концентрации в поглощающем слое атомного пара, то введя эти постоянные величины в значение коэффициента кvи обозначив его k можно записать уравнение как:
Тогда построенный в координатах D и с график будет соответствовать уравнению прямой, проходящей через начало координат.
Упрощенно схему аналитического процесса можно представить в виде следующих этапов.
1. Создают поглощающий слой атомного пара путем введения аэрозоля пробы в пламя или испаряя пробу в графитовом
атомизаторе (атомизация пробы).
2. Через поглощающий слой атомного пара пропускают световой пучок определенной частоты от какого-либо источника света (облучение пробы).
3. Световой поток, прошедший через поглощающий слой, разлагают в спектр и выделяют участок, соответствующий линии
поглощения (выделение из спектра излучения аналитической
линии).
4. Оценивают величину, характеризующую степень поглощения света, прошедшего через поглощающий слой (после получения градуировочной характеристики), т. е. измеряют поглощение аналитической линии.
5. Вычисляют концентрацию определяемого элемента.
АТОМНО-АБСОРБЦИОННЫЙ СПЕКТРОМЕТР
Современный спектрометр представлен на рисунке 1. Свет от источника, испускающего линейчатый спектр определяемого элемента, пропускают через пламя, в которое распыляют раствор анализируемой пробы в виде аэрозоля. С помощью монохроматора выделяют область спектра, соответствующую расположению измеряемой резонансной линии. Излучение выделенной аналитической линии направляют на приемник излучения — фотоэлектрический детектор (обычно ФЭУ) и усилительно-регистрирующую систему, предназначенную для усиления и измерения аналитического сигнала (гальванометр, цифровой вольтаметр или «дисплей», ленточный самописец, телетайп и др.). Интенсивность резонансного излучения измеряют дважды — до распыления анализируемого образца в пламя и в момент его распыления. Разность этих отсчетов служит мерой абсорбции и соответственно мерой определяемого элемента.
Другим способом устранения наложения на выделяемый сигнал излучения атомов, возбуждаемых в пламени (устранения фоновых помех), является использование приема модуляции излучения источника света с частотой 50 или 400 Гц и селективных регистрирующих устройств, настроенных на частоту модуляции.
Рисунок 1 – Схема атомно — абсорбционного спектрометра
1 — линейчатый источник резонансного излучения;
8 — цифропечатающее устройство;
11, 12- ввод окислителя и топлива . соответственно;
Таким образом, единая блок-схема атомно-абсорбционного спектрометра состоит из двух основных частей. Первая служит для превращения анализируемого образца в атомный пар и включает в себя горелку и распылитель со всеми вспомогательными устройствами: газораспределительный блок с приборами для измерения давления и расхода газа, автоматической системой регулирования режима горения и устройствами с автоматическим отключением питания в случае аварийных ситуаций, сюда входит также система газовых коммуникаций блока питания — компрессор для подачи воздуха и баллоны со сжатыми газами. Вторая часть спектрометра служит для выделения и измерения аналитической линии определяемого элемента и включает монохроматор, конденсорные (осветительные) оптические системы и приспособления для модуляции света, источник света, выпрямители — стабилизаторы и СВЧ-генераторы для питания источников света, приемник излучения (ФЭУ), усилительно — регистрирующую систему для усиления и измерения аналитического сигнала, системы управления прибора.
Источники света. В современных приборах в качестве источников света применяют обычно лампы с полыми катодами (ЛПК) или же лампы с СВЧ — возбуждением (СВЧ — лампы), излучающие линейчатый спектр. В качестве вспомогательного источника, излучающего сплошной спектр разряда, используют кварцевые газоразрядные лампы, наполненные дейтерием — так называемый дейтериевый корректор фона.
ЛПК — наиболее распространенный в атомной абсорбции источник света — представляет собой герметичный баллон из молибденового стекла с кварцевым или увиолевым окном, пропускающим ультрафиолетовое излучение (рис. 2). В баллон впаяны два электрода: катод в виде полого цилиндра (иногда закрытого с одной стороны) с отверстием диаметром 2—3 мм, изготовленный из металла (элемента), для определения которого предназначена лампа, и анод произвольной формы. Лампа заполнена инертным газом (аргоном или чаще неоном) под давлением около 10 2 Па и питается сглаженным или пульсирующим током 5—30 мА (сила тока разряда регулируется в зависимости от материала катода и конструктивных особенностей лампы) при выходном напряжении 300—800 В. В этих условиях пары металла, из которого изготовлен катод, интенсивно поступают в плазму разряда и высвечивают соответствующий спектр. Срок службы современных ЛПК составляет 1000— 2000 ч при достаточно высокой стабильности излучения.
 |
Рис 2. Лампа с полым катодом (ЛПК):
В качестве источника резонансного излучения используют также безэлектродные лампы с СВЧ — возбуждением (СВЧ — лампы).
СВЧ — лампа выполнена в виде сферического баллона из плавленного кварца. Внутри баллона, наполненного инертным газом под давлением, находится несколько миллиграммов легколетучего металла (элемента) или его легколетучей соли. Под воздействием высокочастотного электромагнитного поля (для питания этих ламп применяют генераторы, работающие на частотах порядка 2500 Мгц, мощностью около 200 Вт) металл в лампе испаряется и возбужденные атомы концентрируются тонким слоем у поверхности шара, что существенно снижает самопоглощение линий излучения по сравнению с таковыми в других источниках. Использование ламп с СВЧ — возбуждением позволило получать хорошие результаты при определении As, Bi, Sb, Se, Те и Pb, но в общем они менее стабильны по сравнению с ЛПК и не могут конкурировать с ними в простоте и надежности работы.
Системы распылитель — горелка. Основное назначение этих систем заключается в превращении раствора анализируемого вещества в атомный пар. В задачу этой системы входит собственно распыление анализируемого раствора, т. е. превращение его в аэрозоль, отбор фракции аэрозоля с нужным размером капель, смешивание отобранной фракции с компонентами горючей газовой смеси и введение полученной однородной смеси в горелку. Все эти процессы происходят в системах распылитель — горелка с предварительным смешением компонентов в распылительной (конденсационно-смесительной) камере. Последняя выполняет также функции конденсационной камеры для сепарирования капель аэрозоля диаметром более 5 мкм. Поэтому указанные камеры имеют сток для слива конденсата.
В современных спектрометрах наиболее часто применяют системы с распылительными камерами, преимущество которых состоит в том, что благодаря предварительному смешению газов они обеспечивают однородное сгорание топлива и ламинарный характер пламени. Длинные, узкие пламена, для которых характерны высокая стабильность и четко выраженные температурные зоны, позволяют выбрать области с максимальной чувствительностью измерений.
В прямоточных горелках, в которых процесс распыления раствора, а также смешение горючего газа и окислителя происходит после выхода из горелки непосредственно в пламени, потоки газов имеют резко выраженный турбулентный характер. Это приводит к неоднородному горению пламени и тс его нестабильности.
Распылитель является одной из важнейших деталей системы распылитель — горелка. В большинстве современных приборов применяют пневматические распылители инжекционного типа, изготовляемые из -металла, стекла или пластмассы, часто с коррозионно-стойким покрытием внутренней поверхности. Распыление в них происходит под действием воздуха или другого газа — окислителя, подаваемого под давлением 1 -3х10 5 Па. Пневматические распылители бывают концентрические и угловые. В современных приборах используют только наиболее совершенные распылители концентрического типа. С целью получения более мелкодисперсного и однородного аэрозоля применяют ультразвуковые и электростатические распылители, которые, однако, не нашли еще широкого применения.
Для различных горючих газовых смесей используют специально сконструированные горелки (табл. 1). Основное требование к ним состоит в том, чтобы скорость распространения пламени не превышала скорости потока газов через выходное отверстие горелки. При «несоблюдении этого условия возможен проскок пламени внутрь корпуса горелки и оттуда в распылительную камеру с возможным разрушением всего устройства.
Щелевые горелки для газовых смесей довольно однотипны по конструкции и легко заменяются. При работе с воздушно-ацетиленовым пламенем чаще употребляют горелки со щелями 50-0,45 мм и 1000,45 мм, при работе с пламенем оксид азота(1) — ацетилен — горелки из титана со щелями 50-0,55 мм.
Таблица 1 – Типы горелок для атомно – абсорбционного анализа (длина щели горелок первых трёх типов – 10 см, последней – 5 см)
| Тип горелки | Газовая смесь |
| Воздушно-ацетиленовая | Воздух- ацетилен Воздух – водород Аргон – водород |
| Воздушно — пропановая | Оксид азота (1) – пропан Воздух – пропан |
| Многощелевая (горелка Болинга) | Воздух – ацетилен Воздух – пропан Воздух – бутан Аргон — водород |
| Для пламени оксид азота (1) — ацетилен | Оксид азота (1) – ацетилен Воздух – ацетилен |
Рисунок 3 — Схема системы питания горелки и распылителя
1- подача ацетилена (от баллона); 2- подача оксиде азота (I) от баллона; 3 -подача воздуха (от компрессора): 4—воздушный фильтр; 5 -Блок газораспределения; 6 — горелка и смесительная камера; 7 —капилляр распылителя: 6 — стакан с раствором; 9 — сливной шланг; 10 -сосуд для слива
Иногда применяют 3-щелевые горелки 100-0,45 мм, которые позволяют работать с растворами с концентрацией солей до 12% и получать несколько большую чувствительность при использовании воздушно-ацетиленового пламени. Кроме того, эти горелки применяют при работе с воздушно — пропановым пламенем для обеспечения устойчивости горения.
Для поддержания оптимального режима работы системы распылитель — горелка служит блок питания (рис. 3). В нем предусмотрены устройства и приспособления для обеспечения постоянства давления и расхода газов, для автоматического отключения газового потока, для предотвращения аварийных ситуаций и т. д. Блоки питания большинства приборов рассчитаны на возможность использования воздуха и оксида азота в качестве окислителей и ацетилена и водорода в качестве топлива. В блоках обычно используют стандартные баллоны со сжатыми газами.
Система для выделения и измерения аналитической линии. Для выделения резонансной линии в атомно-абсорбционных спектрометрах используют монохроматоры. Оптическая схема монохроматора представлена двумя зачастую симметрично расположенными объективами, в фокусах которых находятся входная и выходная щели и диспергирующее приспособление. Основными характеристиками монохроматора являются разрешающая способность и дисперсия прибора. Разрешение монохроматора должно быть достаточным для разделения линий большинства — источников, излучающих многолинейчатые спектры, и для отделения излучения резонансной линии от молекулярных спектров и сплошного фона пламени.
В качестве диспергирующих элементов в монохроматорах используют призмы и дифракционные решетки. Основное различие между ними заключается в том, что решетки обладают примерно постоянной дисперсией во всей области спектра, а у призм она максимальна в ультрафиолетовой области, но быстро уменьшается с ростом длиныволны. Величина дисперсии зависит от числа штрихов на единицу ширины решетки, спектрального порядка и фокусного расстояния коллиматора.
Большинство приборостроительных фирм используют дифракционные монохроматоры с отражательными решетками и зеркальными объективами, которые обладают достаточно высокой разрешающей способностью и большой светосилой при постоянной дисперсии в спектральном диапазоне.
Оптические схемы, применяемые в атомно-абсорбционных спектрометрах могут быть однолучевыми и двухлучевыми; одно -, двух- и многоканальными. Двухлучевая схема разделяет излучение источника света на два луча, один из которых — основной (он проходит через пламя), а другой — луч сравнения. В приемнике излучения (ФЭУ) и его электроизмерительной схеме определяется отношение этих лучей. Такая схема позволяет устранить влияние нестабильности излучения источника света на полезный сигнал и тем самым повысить точность измерений.
Двухканальная (или многоканальная) оптическая схема позволяет измерять абсорбцию или эмиссию одновременно на двух длинах волн при минимальной затрате пробы. Для этой цели в спектрометре устанавливают два или несколько монохроматоров с таким расчетом, чтобы излучение от источника света, пройдя через пламя, проходило одновременно через все монохроматоры. С помощью монохроматоров выделяют и затем измеряют каждую аналитическую линию. Такой способ можно применять для одновременного определения содержания двух элементов, для одновременного измерения эмиссии и абсорбции. Он может быть использован также для учета фона и при выполнении анализа по методу внутреннего стандарта.
МЕТРОЛОГИЧЕСКАЯ ХАРАКТЕРИСТИКА МЕТОДА
Применение метода атомно-абсорбционного анализа, как и любого другого аналитического метода, для решения проблем охраны окружающей среды требует строгой оценки надежности результатов анализа и их квалифицированной математической обработки. Использование новейших приборов с электронными схемами и первоклассными оптическими системами само по себе еще не гарантирует ни хорошей воспроизводимости, ни правильности результатов определений и требует выявления специфических помех и систематических погрешностей, которые могут достигать больших значений, особенно при работе вблизи предела обнаружения. Важнейшими метрологическими характеристиками атомно-абсорбционного метода являются — предел обнаружения, воспроизводимость и правильность. Предел обнаружения—это числовой критерий, позволяющий объективно судить о возможности обнаружения искомого элемента в пробе. Под пределом обнаружения понимается наименьшая обнаруживаемая в пробе концентрация или наименьшее абсолютное количество искомого элемента, определяемое с заданной доверительной вероятностью. Традиционно в аналитической химии за предел обнаружения принимают минимальное количество элемента, присутствие которого в пробе может быть установлено с доверительной вероятностью 0,95. Это содержание при условии нормального (гауссового) распределения погрешностей численно равно удвоенной величине стандартного отклонения 2S (определенному по серии единичных измерений для концентрации; близкой к уровню холостого опыта, т. е. близкой к пределу обнаружения).
Воспроизводимость результатов анализа — характеристика случайных погрешностей, теория которых (математическая статистика) хорошо разработана. Они зависят от стабильности излучения ламп с полым катодом, от стабильности работы распылительной системы, от стабильности свойств пламени и, наконец, от помех («шумов>) приемников излучения и регистрирующей системы. Поскольку погрешность измерений в атомно-абсорбционном анализе определяется отношением полезный сигнал: шум, а полезный сигнал определяется атомным поглощением, то при уменьшении концентрации определяемого элемента, приводящем к уменьшению поглощения, при сохранении постоянного уровня шумов погрешность определения возрастает. Поэтому воспроизводимость определений при концентрациях, близких к пределу обнаружения, невелика. Относительное стандартное отклонение Sr при содержании 2S равно 0,50 (Sr=Sol2So) =0,50. Более надежным является предел обнаружения, вычисленный по содержанию, численно равному 3S0, что соответствует доверительной вероятности 0,997 и значению Sr, равному 0,33. Таким образом, погрешность Sr дает возможность судить не только о воспроизводимости анализа, но и о значении предела обнаружения. Для многих современных приборов она не превышает 0,01—0,02, поскольку в довольно большом диапазоне концентраций постоянна и близка к минимальной Srмин. В этом диапазоне с минимальным стандартным отклонением — в диапазоне рабочих концентраций — и рекомендуется работать. При оценке же пределов обнаружения более правильно использовать значение стандартного отклонения Sr=Sofc. Оценку воспроизводимости результатов анализа начинают с единичного определения, под которым понимают однократное проведение всей последовательности операций, предусмотренных методикой анализа. Под результатом анализа подразумевают среднее значение результатов нескольких (параллельных) единичных определений, проведенных в одинаковых условиях. Среднее значение концентрации с определяют по формуле
— Ci — результат единичного определения;
Среднее арифметическое ряда параллельных анализов лучше характеризует результат анализа, чем отдельные значения, т. е. отягощено минимальной случайной ошибкой. Получив представительную выборочную совокупность результатов измерений (n >20), стандартное отклонение оценивают по дисперсии:
Таким образом, оценки случайных погрешностей органически связаны с числом параллельных анализов. В соответствии с рекомендациями в качестве меры относительной случайной погрешности принято использовать относительное стандартное отклонение
выраженное в долях определяемой величины. Эта величина в качестве метрологической характеристики анализа удобна тем, что не зависит от единиц измерения определяемой величины, в то время как при выражении результатов анализа в процентах применение коэффициента вариации может привести к путанице [коэффициент вариации W — относительная случайная погрешность, выраженная в %: W= (S/c) 100=Sr , 100%].
Зная предел обнаружения и границы диапазона рабочих концентраций из таблиц можно с помощью соотношения:
—Sr. мни — значение Srв диапазоне концентраций, где его можно считать приблизительно постоянным
с достаточной для практических целей точностью рассчитать значения Sr и S для любой концентрации.
Этой же цели служит приводимое в справочных таблицах значение так называемой характеристической концентрации, т. е. концентрации элемента, соответствующей атомному поглощению 0,0044. Это значение, соответствующее поглощению, равному 1% от интенсивности падающего излучения, т.е. (I — I) / Iо = 0,01, ранее называли аналитической чувствительностью, но в настоящее время ее следует называть «характеристической концентрацией» в отличие от чувствительности, соответствующей наклону калибровочного графика.
Параметры, по которым проводят метрологическую оценку методик анализа, в большинстве случаев определяют экспериментально.
Помимо воспроизводимости, для характеристик аналитического метода или методики чрезвычайно важно понятие правильности анализа, которое характеризуется систематической погрешностью. В свою очередь, систематическая погрешность определяется как статистически значимая разность между средним и действительным значениями содержания определяемого компонента. Выявление, учет и устранение систематических погрешностей осуществляютпа основании детального анализа всех этапов и общей схемы аналитического определения при постановке специальных экспериментов с использованием стандартных образцов. Причем за действительное значение содержания определяемого компонента принимают его содержание в аттестованном во многих лабораториях с применением различных методов природном образце или расчетное содержание в синтетическом образце.
Методические основы АТОМНО- АБСОРБЦИОННОГО АНАЛИЗА
ВЫБОР ОПТИМАЛЬНЫХ УСЛОВИЙ АНАЛИЗА
При выборе оптимальных условий выполнения анализа прежде всего стремятся выполнить два требования: снижение предела обнаружения определяемых элементов и обеспечение ( высокой надежности результатов ^определения. При выборе способа атомизации остановимся на! пламени, которое до сих пор остается удобным, стабильным и экономичным способом получения атомных паров. В течение многих лет практически в любом атомно-абсорбционном спектрометре применяли воздушно-ацетиленовое пламя с предварительным смешением и горелкой камерного типа с щелевой насадкой. И в настоящее время это пламя успешно применяют для определения содержания большинства элементов, не образующих термостойких оксидов. Воздушно-ацетиленовое пламя непригодно для определения металлов с энергией связи металл — кислород выше 5 эВ, например алюминия, тантала, титана, циркония, энергия связи которых соответственно равна 5,98 эВ, 8,4 эВ, 6,9 эВ, 7,8 эВ. Это объясняется необходимостью более высоких температур пламени для элементов с высокой температурой парообразования. Более высокие температуры можно получить при горении смеси кислород — водород и ацетилен—.кислород, но эти смеси имеют высокую скорость горения и трудно поддаются контролю. Поэтому предложенная Виллисом смесь оксид азота (I) — ацетилен сразу получила широкое признание, поскольку наряду с высокой температурой она обладает низкой скоростью распространения пламени и тем самым более безопасна в работе, чем смеси с кислородом.
Таким образом, наиболее применяемыми газовыми смесями являются: воздух — ацетилен и оксид азота (I) — ацетилен.
По количественному соотношению горючего и окислителя газовые смеси могут быть стехиометрическими, обедненными (с содержанием горючего в количестве меньше стехиометрического) и обогащенными (с содержанием горючего в количестве больше стехиометрического). Обедненную смесь называют окислительной, обогащенную — восстановительной.
Газовый состав горючей смеси является основным фактором, определяющим свойства пламени, в том числе его структуру, температуру, стабильность горения и др. Так, пламя оксид азота (1) — ацетилен, получаемое стандартной горелкой, характеризуется наличием трех отчетливых зон: первичной реакционной зоны высотой 2—3 мм светло-голубого цвета, зоны внутреннего конуса, имеющего красноватую окраску и высоту от 0 до 30 мм в зависимости от отношения горючее:окислитель, и голубой диффузионной зоны внешнего конуса. В воздушно – ацетиленовом пламени различают следующие три основные зоны: внутренний конус, тонкую реакционную граничную зону и зону внешнего конуса. Пространственное расположение каждой из зон, их высота, специфичность и активность проходящих в них процессов (дегидратация, испарение, диссоциация химических соединений, окислительно – восстановительные процессы и др.) обусловлены термодинамическими характеристиками пламени. Каждой из зон свойственны свои основные процессы и свои влияющие на них факторы. В связи с этим при выборе оптимальных условий определения того или иного элемента следует обратить внимание не только на рекомендации касающиеся состава газовой смеси, но и на указания об использовании конкретной фотометрируемой зоны пламени.
Неоднородность распределения атомов по высоте пламени обусловлена как скоростью испарения раствора, так и скоростью реакции диссоциации присутствующих в пламени соединений (гидрооксидов, оксидов и др.). То есть время жизни свободных атомов различных элементов зависит от степени их сродства к кислороду и некоторым радикалам углеводородных пламен. Для элементов с высокой степенью сродства к кислороду зона максимальной абсорбции в пламени ограничена небольшим участком с наибольшей температурой над вершиной внутреннего конуса – 6-12 мм над основанием горелки.
В реальных условиях проведения анализа следует учитывать одновременно многие факторы: состав газовой смеси, расход окислителя и горючего, неоднородность структуры пламен, ток питания лампы и многие другие.
Большое значение при выборе оптимальных условий выполнения анализа имеет также правильный выбор режима работы источников резонансного излучения, в качестве которых чаще всего используют лампы с полым катодом. Интенсивность излучения лампы, которая может быть использована для снижения уровня шумов фототока, обычно возрастает при увеличении силы тока питания лампы. Однако с увеличением силы тока ламп иногда наблюдается уменьшение аналитического сигнала, а для таких легколетучих металлов, как цинк, магний и кадмий, появляется эффект самообращения линий, приводящий к ослаблению интенсивности центральных участков этих резонансных линий. Поэтому для источников излучения линий этих металлов рабочие токи ограничены для ламп с полым катодом до 15—20 мА и для высокочастотных ламп — до 160—180 мА.
Аналогично большинству количественных аналитических методов в методе атомно-абсорбционной спектроскопии предполагается перед измерениями получение градуировочных характеристик, т. е. установление взаимооднозначного соответствия между концентрациями определяемого элемента в стандартных растворах (растворах сравнения) и ценой деления регистрирующего прибора в конкретных условиях опыта. Операцию градуировки (калибровки) измерений проводят всякий раз перед началом работы и после любого перерыва в работе, вызванного гашением пламени, юстировкой лампы и т. д. В качестве растворов сравнения используют в основном синтетические растворы с надежно установленным содержанием определяемого элемента. Поскольку наиболее часто атомно-абсорбцнонный метод применяют для определения следовых количеств элементов в водных растворах, растворы сравнения (эталоны) также готовят обычно путем растворения металла или его соли в кислоте, разбавляя затем до определенного объема водой или слабым раствором кислоты.
Для выяснения хода калибровочной кривой основного раствора сравнения по мере надобности готовят серию разбавленных рабочих растворов, поглощательная способность которых соответствовала бы оптимальному интервалу измерений (например, 0; 0,2; 0,4; 0,6 единиц поглощательной способности). При существенных матричных эффектах состав стандартных растворов должен приближаться к составу проб, в противном случае следует использовать метод стандартных добавок. Для сильно разбавленных растворов матричные эффекты могут быть малозначимыми.
источник
| Название: Атомно-адсорбционный спектрохимический анализ тяжелых металлов в почве Раздел: Рефераты по экологии Тип: курсовая работа Добавлен 15:07:28 12 января 2010 Похожие работы Просмотров: 4864 Комментариев: 14 Оценило: 3 человек Средний балл: 5 Оценка: неизвестно Скачать | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Элемент | Концентрация |
| Цинк | 0,1 — 8,0 мг/дм 3 |
| Хром | 0,001 — 0,1 мг/дм 3 |
| Марганец | 0,0003 — 0,050 мг/дм 3 |
| Медь | 0,0005 — 0,070 мг/дм 3 |
| Железо | 0,005 — 0,060 мг/дм 3 |
Объем дозируемой в атомизатор пробы — от 5 до 40 мм 3 .
При анализе проб воды с массовой концентрацией металлов, превышающей верхнюю границу диапазона, допускается разбавление пробы бидистиллированной (деионизованной) водой, но не более чем в 100 раз. (Zn, Cr), не более чем в 1000 раз (Mn, Cu, Fe)
Метод измерения основан на резонансном поглощении света свободными атомами металлов, возникающем при пропускании света через слой атомного пара в графитовой печи атомно-абсорбционного спектрометра «МГА-915». Содержание металлов определяется величиной интегрального аналитического сигнала и рассчитывается по предварительно установленной градуировочной зависимости.
Методика обеспечивает выполнение измерений с погрешностью, не превышающей величин, указанных в табл.2.
Значения характеристики погрешности измерений для доверительной вероятности Р=0,95
| Определяемый элемент | Воды природные | |
| Диапазон измеряемых массовых концентраций, мг/дм 3 | Характеристика погрешности измерений, ±δ, % | |
| Марганец | 0,0003-0,005 0,005-0,020 0,020-0,050 | 35 20 15 |
| Медь | 0,0005-0,010 0,010-0,025 0,025-0,070 | 50 25 15 |
| Цинк | 0,1-0,4 0,4-0,8 | 35 18 |
| Железо | 0,005-0,010 0,010-0,060 | 35 20 |
| Хром | 0,001-0,005 0,005-0,1 | 40 20 |
| Спектрометр атомно-абсорбционный «МГА-915» | ТУ 4434-915-205016233-98 |
| Весы лабораторные общего назначения 2-го класса точности, например ВЛР-200 | ГОСТ 24104 – 88 |
| Меры массы | ГОСТ 7328 – 82 |
| Мерные колбы 2-1000-2, 2-100-2, 2-50-2 | ГОСТ 1770 – 74 |
| Пипетки градуированные 2-го класса точности вместимостью 1, 2 , 5 и 10 см 3 | ГОСТ 29227 — 91 |
| Дозатор пипеточный одноканальный переменного объема 5-50 мм 3 . Погрешность измерения – не более ±5 % | ТУ 9452-001-33189998-95 |
| Государственные стандартные образцы состава раствора определяемых ионов (1 мг/см 3 , погрешность аттестованного значения ±1 %): | ГСО 7266 — 96 |
| • марганца | ГСО 7266 — 96 |
| • меди | ГСО 7255 — 96 |
| • железа | ГСО 7254 — 96 |
| • цинка | ГСО 7256 — 96 |
| • хром | ГСО 7768 — 2000 |
Допускается использование средств измерений и стандартных образцов с аналогичными или лучшими метрологическими характеристиками. Средства измерений должны быть поверены в установленные сроки.
Общие требования к отбору проб по ГОСТ Р 51592-2000. Отбор проб природной воды производится по ГОСТ 17.1.5.05-85, сточной воды по НВН 33.5.3.01-85. Объем отбираемой пробы составляет не менее 50 см 3 .К пробе добавляют 3 см 3 концентрированной азотной кислоты на 1 дм 3 пробы и хранят в посуде из полиэтилена, полипропилена или фторопласта. Пробы перед анализом фильтруют через бумажный фильтр «белая лента» или пористый фильтр с диаметром пор 0,45 мкм. При фильтровании первые порции фильтрата (не менее 5 см 3 ) следует отбросить. Посуду, предназначенную для отбора проб и хранения проб, промывают раствором азотной кислоты, а затем дистиллированной и бидистиллированной (ионизированной) водой. Срок хранения законсервированной пробы — 3 дня.
Для каждого раствора необходимо использовать свою пипетку. Раствор из колбы наливают в стаканчик и из него набирают пипетку. Запрещается погружать пипетку во весь объем раствора во избежание загрязнения.
Рекомендуется иметь отдельный набор посуды для приготовления растворов каждого элемента.
Все растворы готовят на бидистиллированной (деионизованной) воде.
Раствор азотной кислоты, объемная доля 2%
В коническую колбу помещают 300-400 см 3 бидистиллированной деионизованной воды, осторожно, при перемешивании вливают 10 см 3 концентрированной азотной кислоты, доводят до 500 см 3 бидистиллированной водой и перемешивают. Раствор устойчив при хранении в закрытом сосуде из полиэтилена, полипропилена или фторопласта длительное время. Контроль качества раствора азотной кислоты проводят ежедневно перед началом работы. Процедура контроля состоит в измерении массовой концентрации элементов в соответствии с п.7 методики. Если измеренное значение превышает 30% нижней границы диапазона измерения элемента (п.1),то раствор необходимо приготовить заново.
Приготовление градуировочных растворов
Приготовление рабочих растворов массовой концентрации 100мг/дм 3
В мерную колбу вместимостью 50 см 3 помещают при помощи пипетки 5 см 3 государственного стандартного образца состава раствора соответствующего иона, доводят до метки 2 % -ным раствором азотной кислоты и перемешивают. Раствор устойчив при хранении в полиэтиленовой посуде в течение 1 месяца.
Приготовление рабочих растворов массовой концентрации 2мг/дм 3
В мерную колбу вместимостью 100 см 3 помещают при помощи пипетки 2 см 3 рабочего раствора массовой концентрации 100 мг/дм 3 , приготовленного по п. 6.2.2.1, доводят до метки 2%-ным раствором азотной кислоты и перемешивают. Раствор устойчив при хранении в полиэтиленовой посуде в течении 2 недель.
Приготовление рабочих растворов массовой концентрации 1мг/дм 3
В мерную колбу вместимостью 100 см 3 помещают при помощи пипетки 1 см 3 рабочего раствора массовой концентрации 100 мг/дм 3 , приготовленного по п. 6.2.2.1, доводят до метки 2%-ным раствором азотной кислоты и перемешивают. Раствор устойчив при хранении в полиэтиленовой посуде в течении 2 недель.
Приготовление рабочих растворов массовой концентрации 20мкг/дм 3
В мерную колбу вместимостью 100 см 3 помещают при помощи пипетки 2 см 3 рабочего раствора массовой концентрации 1 мг/дм 3 , приготовленного по п. 6.2.2.3, доводят до метки 2%-ным раствором азотной кислоты и перемешивают. Раствор используют свежеприготовленным.
Приготовление рабочих растворов массовой концентрации 10мкг/дм 3
В мерную колбу вместимостью 100 см 3 помещают при помощи пипетки 1 см 3 рабочего раствора массовой концентрации 1 мг/дм 3 , приготовленного по п. 6.2.2.3, доводят до метки 2%-ным раствором азотной кислоты и перемешивают. Раствор используют свежеприготовленным.
Приготовление рабочих растворов массовой концентрации 1мкг/дм 3
В мерную колбу вместимостью 50 см 3 помещают при помощи пипетки 5 см 3 рабочего раствора массовой концентрации 10 мкг/дм 3 , приготовленного по п. 6.2.2.5, доводят до метки 2%-ным раствором азотной кислоты и перемешивают. Раствор используют свежеприготовленным.
Для построения градуировочной зависимости аналитического сигнала от массы элемента в графитовую печь атомизатора вводят дозатором необходимый объем (от 5 до 40 мм 3 ) градуировочных растворов соответствующего элемента. Диапазоны построения градуировочной зависимости приведены в таблице 2. Необходимо использовать не менее 5 точек в указанном в таблице 2 диапазоне массы. При построении градуировочной зависимости следует начинать с меньших значений массы элемента и от них переходить к более высоким. В таблице 3 приведены рекомендуемые для внесения объемы градуировочных растворов.
Рекомендуемые режимы обработки градуировочных растворов и проб приведены в таблице 4.
Измерение с каждой массой элемента проводят 5 раз в соответствии с Руководством по эксплуатации спектрометра (далее РЭ) и рассчитывают среднее арифметическое значение полученных значений. Затем запускают процедуру «Ручная градуировка» и вводят с клавиатуры компьютера массу элемента (в пиктограммах) и соответствующие им величины средних значений аналитического сигнала. Полученную градуировочную зависимость можно просмотреть в режиме «Градуировка»/«Просмотр».
Диапазоны построения градуировочных зависимостей.
| Элемент | Диапазон измерения, мг/дм 3 | Диапазон масс, пг |
| Марганец | От 0.0003 до 0.050 включительно | 10 — 400 |
| Медь | От 0.0005 до 0.070 включительно | 20 — 600 |
| Железо | От 0.005 до 0.060 включительно | 40 — 600 |
| Цинк | От 0.1 до 8.0 включительно | 5000 — 80000 |
| Хром | От 0.001 до 0.1 включительно | 40 — 600 |
Рекомендуемые способы внесения элемента в атомизатор
| Масса, пг | Концентрация градуировочного раствора, мкг/дм 3 | Объем градуировочного раствора, мм 3 |
| 10 | 1.0 | 10 |
| 20 | 1.0 | 20 |
| 40 | 1.0 | 40 |
| 100 | 10.0 | 10 |
| 200 | 10.0 | 20 |
| 400 | 10.0 | 40 |
| 600 | 20.0 | 30 |
| 800 | 20.0 | 40 |
При высоких значениях массы элемента может наблюдаться отклонение градуировочной зависимости от линейной. В этом случае рекомендуется ограничиться более узким, чем в указано в табл. 2 интервалом массы определяемого элемента для построения градуировочной зависимости.
Контроль стабильности градуировочной зависимости
Контроль стабильности градуировочной зависимости состоит в проведении не менее двух параллельных измерений массовой концентрации растворов, заново приготовленных по п. 6.2.2, перед началом работы, и после анализа 15-20 проб.
Градуировка признается стабильной, если расхождение между заданным и измеренным значением концентраций не превышает 15% от заданного значения. В этом случае процесс измерений признается подконтрольным, и результаты измерений массовой концентрации элемента в пробах за период между двумя последовательными процедурами контроля стабильности градуировочной характеристики принимаются в качестве окончательных результатов.
При несоответствии полученных результатов указанному нормативу процесс градуировки необходимо повторить.
Вводят дозатором в графитовую печь атомизатора от 5 до 40 мм 3 анализируемой пробы (в зависимости от ожидаемого содержания) и производят измерение в соответствии с выбранным режимом работы (таб. 4). Режимы при измерении градуировочных растворов и проб (за исключением стадии пиролиза) должны совпадать. Температура и продолжительность пиролиза зависят в первую очередь от матричного состава пробы. При анализе сравнительно чистых или разбавленных сточных вод режим пиролиза можно не использовать.
Порядок проведения измерений осуществляется в соответствии с Руководством по эксплуатации спектрометра. Объем дозированной пробы вводится с клавиатуры компьютера по запросу программы. После завершения измерения на дисплей компьютера выводится величина интегрального аналитического сигнала, масса и концентрация определяемого компонента. Полученные данные автоматически протоколируются. Анализ пробы осуществляется минимум 2 раза.
Если измеренное значение массы элемента в пробе выходит за область линейности градуировочной характеристики, то пробу необходимо разбавить бидистиллированной (деионизованной) водой, предварительно проверенной на наличие примеси определяемого элемента. Затем разбавленную пробу анализируют как описано выше. Коэффициент разбавления пробы Q вычисляют по формуле[8-16]
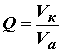 | (1) | |
| Vа | аликвотная порция исходной пробы, взятая для разбавления, см 3 . | |
Разработка методического обеспечения атомного оптического спектрального анализа почв и биологических материалов для экологической экспертизы на токсичные металлы[23]
Разработан простой и экспрессный способ кислотного разложения почв и биологических объектов при воздействии ультразвуком для определения ртути, свинца и других тяжелых металлов из одного раствора. На этой основе предложены новые экспрессные методики последовательного атомно-абсорбционного определения ртути на ртутном анализаторе “Юлия-2” и свинца, цинка, меди – на пламенном атомно-абсорбционном спектрометре “AAS-1“ в пламени пропан-воздух.
Проблему повышения чувствительности определения свинца и многоэлементного анализа решали спектрографическим методом на дифракционном спектрографе ДФС 8–1 после упаривания раствора пробы с углеродным коллектором и введением сухого концентрата в дуговой разряд воздушной струей на спектральной установке “Полюс –2”.
Методики были применены к анализу почв, донных отложений и волос человека.
Было изучено распределение Hg , Pb, Zn , Cu в почвах и в выделенных из них гуминовых кислотах по разрезу учебного полигона ИГУ, разрабатываемого совместными усилиями преподавателей и студентов биолого-почвенного, географического и химического факультетов.
Проведена экспертиза почв Усольского района, находящихся под несанкционированной свалкой УПО “Химпром”, и было выявлено загрязнение по натрию, бору и хрому.
Представлены содержания Hg , Pb и Zn в волосах детей 6 и 12 лет Академгородка г.Иркутска, которые были исследованы с целью медико-экологического мониторинга в рамках довузовской программы со школьниками “Экология и здоровье”.
Выполнен аттестационный анализ разработанных в Институте геохимии СО РАН стандартных образцов состава донных отложений оз. Байкал БИЛ–1 и БИЛ–2 на ртуть. Определены содержания исследуемых металлов в донных отложениях Братского водохранилища. Подтверждены данные о повышении содержания ртути (ртутная проблема водохранилищ).
Квантовохимическое моделирование и исследование пиролиза серусодержащих хелатов меди, кадмия свинца атомно-абсорбционным методом[22]
Серусодержащие комплексообразующие модификаторы матрицы позволяют устранить влияние основы, улучшить воспроизводимость и снизить предел обнаружения при атомно-абсорбционном (АА) определении меди, кадмия, свинца и др. элементов в сложных объектах. Эффективность добавки определяется высокотемпературным процессом пиролиза комплексов в графитовых печах на доатомизационных станциях. АА методом установлено влияние на аналитический сигнал свинца (II), меди (II) и кадмия (II) добавок диэтилдитиокарбамата натрия и бензтиазолилмеркаптометилсульфида. Вышеуказанные хелаты были выделены в твердом виде, подтвержден их состав и дериватографическим методом установлено, что их разложение носит ступенчатый характер и завершается к 500 С. Квантовохимическое моделирование позволило оценить вероятный путь пиролиза хелатов. Поиск оптимальной конфигурации ядерного остова выполнялся в приближении Девидона-Флетчера. расчет электронной структуры выполнен в полуэмпирическом приближении МО ЛКАО. Использован комплекс квантовохимических программ МОРАС 6. Для диэтилдитиокарбамата свинца с точки зрения величины теплоты возможного процесса наиболее вероятным представляется радикальный пиролиз с разрывом связи Pb–S и образованием связанного с сухим углеродным остатком свободного радикала, стабилизированного системами конденсированных колец образующихся при карбонизации углеродсодержащих соединений. Другими продуктами пиролиза являются промежуточный хелат с меньшим, по сравнению с исходным, содержанием диэтилдитиокарбамата и, вероятно, сероводород. Аналогично протекает пиролиз и бензтиазолилмеркаптометилсульфида меди. Диэтилдитиокарбамат кадмия при разложении также образует свободный радикал, сульфид кадмия и газообразные продукты, состав которых существенно изменяется в зависимости от условий нагрева атомизатора, содержания кислорода в инертном газе, от состояния поверхности печи и т.д.
Предложенные модели пиролиза экспериментально подтверждены дериватографическим, рентгенофазовым методами, ЭПР-спектроскопией, исследованием молекулярных спектров АА методом. Обнаруженная общая стадия высокотемпературного разложения серусодержащих комплексов с образованием свободных радикалов изменяет функцию испарения и переноса металлов в графитовой печи, что способствует термостабилизации и объясняет изменение метрологических характеристик АА методик в присутствии добавки.
Изучение поведения токсичных элементов в природных средах методом атомной абсорбции.[21]
Проведено комплексное исследование загрязнения воздушного пространства, снега, почв, водных источников на основе атомно-абсорбционного определения токсичных элементов в каменных углях, золе уноса Беловского района Кемеровской области.
В качестве базовой информации были использованы результаты аналитических исследований 100 образцов твёрдых веществ в снежном покрове в период максимального влагозапаса, твёрдых веществ, содержащихся в приземном воздухе, 48 образцов воды из питьевых источников, 130 образцов почв, 30 образцов листьев древесных растений, 70 образцов растительных кормов сельскохозяйственных животных, 120 образцов пищевых продуктов человека. Всего было выполнено более 9000 элементо-определений с целью обнаружения в них Cu, Be, Hg, Zn, Cd, Pb, As, Cr, Mn, Fe, Co, Ni.
Определение химических элементов проводили на спектрофотометрах Сатурн-2М и ААS-1N, с пределами обнаружения в мкг/мл: 0,002 для Ве и Аs; 005 для Mn и Cu; 0,01 – Zn, Cd ,Cr; 0,02 – Ni; 0,05 – Cr; 0,1 для Hg.
Осуществлён анализ проблемы загрязнений воздушного бассейна теплоэнергетическими объектами. Обосновано приоритетное значение твёрдых атмосферных выбросов теплоэнергетических объектов для местных экосистем и популяций человека. Изучен состав токсичных химических веществ в кузбасских каменных углях и рассчитан их баланс в процессе сжигания твёрдого топлива на Беловской ГРЭС. Установлено, что на ГРЭС с золой уноса в воздушный бассейн поступает 3-39% массы токсичных веществ, а 61-97 % этих веществ ассоциируется с золошлаковой смесью и сбрасывается в золоотвал. Техногенная химическая нагрузка на изучаемую территорию определена по содержанию вредных элементов в снежном покрове в период максимального влагозапаса. Эти данные использованы для расчёта доз химических токсикантов, поступающих в организм местных жителей.
Определение форм тяжелых металлов в снежном покрове после экстракции тиопирином[20]
В работе представлены экспериментальные результаты снегосъемки 2002-2003 гг. на опорных площадках промышленного центра и в области его влияния. Методом атомно-абсорбционного анализа водной и кислотной вытяжек из почвогрунтов, а также экстрактов из твердой компоненты снега и образцов после их мокрого озоления количественно определены различные формы свинца. В работе было показано, что оптимальное количество тиопирина для извлечения катионов металлов составляло 4*10 -3 моль при фиксированном количестве трихлоруксусной кислоты 2*10 -2 моль.Тиопирин хорошо растворим в воде при нагревании. Данный реагент склонен к протонизации с образованием катионной формы: Тиопирин и его аналоги обладают сочетанием ярко выраженных свойств ароматических соединений и алифатических аминов. Это объясняется наличием в гетероцикле секстета p — электронов, т.к. в молекуле тиопирина неподеленные p — электроны атомов азота включаются в общую p — электронную систему тиопиринового цикла. Вследствие этого атомы азота теряют свои электрондонорные свойства, а атом серы получает значительный отрицательный заряд. Таким образом, донорным атомом служит атом серы. Тиопирин является монодентным лигандом, взаимодействующим с неорганическими катионами Pb(II), хемосорбированными на частицах твердого аэрозоля и пылевых выпадений в виде неорганических солей с анионом X 2 — ( сульфид, сульфат, карбонат). При этом образование комплекса со свинцом зависит от pH
При этом протоны водорода могут встраиваться во внутреннюю сферу комплекса.
Таким образом установлено, что на поверхности твердых частиц снежной массы преобладает на 80 — 90% неорганический свинец, на органические формы свинца приходится 3 — 9 %, в том числе 2 — 5 % антропогенного происхождения. Метод определения соотношения форм предлагается использовать для экологического контроля за состоянием атмосферы и снежного покрова в зимний лей.
Спектрометрические характеристики растворов гумуса, исследование их комплексообразования с металлами[19]
Исследование комплексообразующих (КО) свойств гуминовых веществ (ГВ) проводят с целью их использования для уменьшения токсичности тяжёлых металлов в природных объектах, что представляет интерес для агрохимии, почвоведения и курортологии. Сложный состав ГВ, большая зависимость их строения от географического положения объекта, из которого они выделены (почвы, уголь, торф, сапропель, вода), не позволяют распространять результаты исследований и методологические подходы ко всем ГВ.
Методом ААС установлено, что в образцах ГВ содержатся: К, Са, Mg от 1 до 10%; Fe, Zn, Na от 10 -2 до 10 -1 %; остальные металлы от 10 -4 до 10 -3 %. Определяющее влияние на процесс КО ГВ с Cu 2+ и Pb 2+ оказывает присутствие металлов первой группы. Обработка ГВ 0.1н HCl в течение 12 часов ведет к потере К и Са. Использование Н-формы ГВ более перспективно для КО с металлами.
Электронные спектры водных растворов ГВ и их растворов с Cu 2+ , Pb 2+ в ацетатно-аммиачных буферах (при рН 5, 6, 7, 8) показали, что растворы ГВ в воде имеют интенсивные полосы поглощения, например λ 1 max =207-208 нм, которые в буферных растворах смещаются в длинноволновую область до λ 2 max =235 нм. КО ГВ с ионами металлов имеет сложный характер. Поэтому для изучения КО были также использованы ИК-спектры. Образование связи карбоксилат-ион-металл устанавливали по исчезновению полосы поглощения 1720 см -1 и появлению полосы 1580 см -1 .
На основе полученных данных можно прогнозировать поведение ионов металлов в природных объектах, в том числе при содержаниях на уровне ПДК.
Термическая атомно-абсорбционная спектроскопия как метод диагностики форм нахождения тяжелых металлов в объектах окружающей среды и минералах[18]
Термический атомно-абсорбционный анализ (ТАА) является совмещением термического и атомно-абсорбционного анализа. Метод основывается на одновременной регистрации сигнала абсорбционности от спектрометра и температуры пробы. При этом диагностическими характеристиками являются температурные параметры выхода элементов, зависящие от форм присутствия данных элементов в минеральном веществе. Предварительно производится калибровка этих параметров по синтетическим минералам с заданными формами нахождения элемента. В настоящее время метод разработан для анализа форм ртути, кадмия, свинца, цезия. Данный метод позволяет решать широкий спектр задач геохимии, неорганической химии и охраны окружающей среды.
Проведены системные исследования форм нахождения тяжелых металлов (Hg и Cd) в осадках водохранилищ Ангарского каскада, донных отложений озера Байкал и акватории Охотского, Берингова и Японского морей. Установлено, что основной поток ртути в озеро Байкал и его донные отложения направлен из атмосферы. В донных отложениях Братского водохранилища обнаружено широкое развитие сорбционных форм кадмия, что необходимо учитывать при мониторинге кадмиевого загрязнения на территории Иркутской области, так как он может быть токсичным для органических объектов водохранилищ.
ТАА позволил показать различие в механизмах поглощения микроэлементов при исследовании образования устойчивых форм Cd и Hg в пирротине и галените (в условиях гидротермального синтеза при повышенных температурах и давлениях). Это стало возможным благодаря сопряжению методов ТАА и спектроскопии поверхности. Большое влияние на механизм захвата оказывают дефекты структуры исходных минералов. Особое значение имеет механизм с участием так называемых «неавтономных фаз», так как они более устойчивые по сравнению с поверхностными комплексами.
3. СОВРЕМЕННОЕ ОБОРУДОВАНИЕ
Атомно-абсорбционные и эмиссионные спектрометры
Атомно-абсорбционный спектральный анализ основан на селективном поглощении УФ — или видимого излучения атомами газа.
Для перевода пробы в газообразное атомарное состояние применяются два вида устройств атомизации — пламенные и электротермические.
В качестве источника излучения обычно применяют лампу с полым катодом из определяемого металла. Интервал длин волн спектральной линии, испускаемой источником сета, и линии поглощения того же самого элемента в пламени очень узок, поэтому поглощение других элементов практически не сказывается на результатах анализа.
Атомно-абсорбционные элементные анализаторы относятся к современным селективным, высокопроизводительным и точным приборам, которые позволяют анализировать до 70 элементов в пробе с чувствительностью в интервале 10 -4 -10 -9 % масс. Недостатками этого вида анализа являются необходимость использования горючих газов, невозможность одновременного определения в пробе нескольких элементов.
В настоящее время известно несколько модификаций средств измерений, основанных на принципе атомной абсорбции, выпускаемых отечественными фирмами: — «Спектр — 5М»: ширина спектрального диапазона прибора — от 190 до 800 нм, время одного измерения — 1 мин, «КВАНТ — АФА», «КВАНТ — Z. ЭТА», «МГА — 915».
В настоящее время метод атомной абсорбции считается одним из самых селективных, производительных, экспрессных, точных и одновременно сравнительно дешевых (7 — 15 тыс. $).
Вариантом атомной спектроскопии является атомно-эмиссионная спектроскопия, отличающаяся от атомно-абсорбционной обратным способом регистрации — по оптическому спектру испускания возбужденных атомов.
В этом варианте атомизатор и источник возбуждения совпадают, что несколько упрощает конструкцию. Наиболее перспективным считается вариант с индуктивно связанной плазмой (ИСП), не уступающей по чувствительности атомно-абсорбционным атомизаторам, но имеющий в 10 — 100 раз более широкий диапазон определяемых содержаний. При этом атомно-эмиссионные анализаторы позволяют одновременно определять в пробе несколько элементов, но к сожалению, уступают атомно-абсорбционным спектрометрам по воспроизводимости и по селективности.
Среди имеющихся на рынке наиболее известны приборы серии «ЭРИДАН-500». Будучи основанными на ИСП, эти эмиссионные спектрометры позволяют проводить элементный анализ практически любых веществ, в том числе чистых металлов и примесей в них, сплавов и сталей, порошковых (в том числе почв) и жидких проб (в том числе после поглощения из воздуха), продуктов питания, медицинских проб с высокой точностью (1- 20 %). Пределы обнаружения Cr, Al, Hg, As, Ni, Pb составляют 1 — 20 мкг/л. Стоимость данной модификации составляет 22 000 $.
Атомно-абсорбционный анализ (ААА) является одним из наиболее распространенных методов аналитической химии. Предварительная подготовка анализируемой пробы аналогична этой операции в пламенной фотометрии: перевод пробы в раствор, распыление и подача аэрозолей в пламя. Растворитель испаряется, соли разлагаются, а металлы переходят в парообразное состояние, при котором они способны поглощать излучение той длины волны, которую могли бы сами излучать при более высоких температурах. Луч света от лампы полого катода, излучающий дуговой спектр определяемого элемента, направляется через пламя на щель спектрометра, с помощью которого выделяется аналитическая спектральная линия и измеряется степень поглощения ее интенсивности парами определяемого элемента.
Атомно-абсорбционные спектрометры AAnalyst 200 и 400 устанавливают новые стандарты в области анализа элементного состава материалов. Это первые серийные приборы с реальной двухлучевой Эшеле — оптической системой. AAnalyst 200, имеющий встроенную систему управления с графическим интерфейсом и сенсорным экраном, кардинальным образом меняет представления о методе атомной абсорбции с пламенной атомизацией. Никогда ранее этот метод не был столь прост и не обладал такими возможностями! AAnalyst 400 — атомно-абсорбционный спектрометр, дающий полную автоматизацию пламенного и печного вариантов АА при безупречных параметрах по доступной цене.
Атомно-абсорбционный спектрометр с электротермической атомизацией и Зеемановской коррекцией неселективного поглощения «МГА-915» предназначен для измерения содержания элементов (Ag, Al, As, Au, Ba, Be, Bi, Cd, Co, Cr, Cu, Fe, Hg, Mn, Mo, Ni, Pb, Pd, Pt, Rh, Ru, Se, Sn, Sb, Sr, Ti, V, Zn и др.) в широком круге объектов: различных типах вод (питьевые, природные, сточные, морские), атмосферном воздухе, почвах, донных отложениях и осадках сточных вод, пищевых продуктах и сырье (в том числе в напитках), биологических тканях и жидкостях (кровь, моча), продуктах нефтехимического производства, а также металлах и сплавах и иных объектах. Наибольшей эффективностью данный прибор обладает при анализе проб со сложным матричным составом: морские воды, кровь, моча.
Спектрометр может комплектоваться автосемплером, ртутно-гидридой приставкой. В качестве источников света используются лампы с полым катодом, а также высокоинтенсивные безэлектродные разрядные лампы собственного производства.
Спектрометр «МГА-915» обладает следующими характеристиками:
Универсальность и селективность. Высокая селективность связана с использованием высокоэффективного варианта селективного атомно-абсорбционного анализа – Зеемановской модуляционной поляризационной спектрометрии. Анализатор «МГА-915», благодаря своей высокой селективности, позволяет определять содержание широкого круга элементов в пробах самого разного состава – без или с минимальной пробоподготовкой. ААС с ЭТА и зеемановским корректором неселективного поглощения во всем мире признан в качестве «референтного метода» при определении малых содержаний элементах в пробах сложного состава.
Высокая чувствительность. Пределы обнаружения элементов на уровне лучших атомно-абсорбционных спектрометров, предлагаемых на рынке аналитического оборудования.
Автоматизация измерений. «МГА-915» является полным автоматом с автоматической сменой источников излучения и установкой соответствующих резонансных линий, присутствует турель на 6 ламп (компьютерная перестройка с одного элемента на другой без необходимости ручной регулировки.
Применяется в экологии, геологоразведке, контроле технологических процессов, производственной санитарии, научных исследованиях.
· измерение содержания различных элементов в воде, почве, донных отложениях, атмосферном воздухе, а также тканях растительного и животного происхождения.
· экспресс-анализ и непрерывный контроль состава веществ в технологических процессах;
входной контроль, контроль готовой продукции
· анализ тканей и жидкостей биологического происхождения (кровь, моча, волосы и др.)
· идентификация примесей и следовых количеств элементов.
· корма, кровь, продукты животноводства.
Контролирующие и сертифицирующие лаборатории: анализ пищевых продуктов и кормов, анализ сточных, природных, питьевых вод.
Атомно-абсорбционный спектрофотометр «Спираль-17»
Спектрофотометр СПИРАЛЬ-17 предназначен для определения концентрации токсичных металлов в питьевой, природных и сточных водах, пищевых продуктах, почве, воздухе, растениях и других объектах.
Новая модель спектрофотометра с вольфрамовым спиральным атомизатором отличается от первого серийного прибора СПИРАЛЬ-14, нашедшего применение в более чем 80 лабораториях Госсанэпиднадзора, водоканалов, охраны окружающей среды, промышленных предприятий и институтов, улучшенными аналитическими и эксплуатационными характеристиками, повышенной надежностью в работе.
Спектрофотометр СПИРАЛЬ-17 внесен в Госреестр средств измерений, имеет сертификат и допущен к применению в Российской Федерации.
Основные преимущества спектрофотометра СПИРАЛЬ-17:
· спиральный вольфрамовый атомизатор, имеющий чувствительность на уровне графитового, длительный срок службы, малое энергопотребление и отсутствие водяного охлаждения;
· полная автоматизация процесса анализа, обработка результатов измерений и управление работой прибора от ПЭВМ типа IBM PC;
· отсутствие горючих газов, большая в 100-1000 раз чувствительность определения большинства элементов в сравнении с пламенными атомно-абсорбционными спектрофотометрами;
· возможность определения широкого круга элементов на уровне предельно допустимых концентраций (ПДК) и ниже;
· меньшая стоимость прибора в сравнении с отечественными и зарубежными аналогами.
По заявкам потребителей разрабатываются с аттестацией в Госстандарте, методики анализа других, интересующих заказчиков объектов.
Изготовитель обеспечивает проведение пуско-наладочных работ, гарантийное и сервисное обслуживание, обучение и консультации, поставку расходуемых материалов.
Основные параметры и характеристики спектрофотометра
| Спектральный диапазон, нм | от 200 до 600 |
| Диапазон измерения оптической плотности, | Б 0-1,5 |
| Корректор неселективного поглощения | дейтериевый |
| Объем отбираемой пробы, мкл | 6 |
| Время измерительного цикла, с | 60-90 |
| Расход защитного газа (аргон), л/мин | 0,5-1 |
| Среднее число рабочих циклов атомизатора, шт. | 1500 |
| Потребляемая мощность, В*А | не более 300 |
| Масса, кг | не более 50 |
| Габаритные размеры, мм, не более: | 680*350*610 |
Для более эффективного использования прибора разработаны, аттестованы в Госстандарте и поставляются потребителям методики определения:
· Ag, AI, Cd, Co, Cr, Cu, Fe, Mn, Ni, Pb, Zn в питьевой и природной воде;
· AI, Cd, Cr, Cu, Fe, Mn, Ni, Pb, Sn, Zn в сточных водах;
· AI, Cd, Co, Cr, Cu, Fe, Mn, Ni, Pb, Zn в воздухе рабочей зоны и промвыбросах;
· Cd, Cu, Pb, Zn в зерне, муке и хлебобулочных изделиях;
· Cd, Cu, Fe, Pb, Zn в пищевом спирте, водке и вине. [24]
Пределы обнаружения и ПДК в питьевой воде для некоторых элементов (в мг/л)
| Элемент | Предел обнаружения | ПДК | Элемент | Предел обнаружения | ПДК |
| Аg | 0,0001 | 0,05 | Cu | 0,0001 | 1,0 |
| Al | 0,0003 | 0,5 | Fe | 0,0004 | 0,3 |
| Bi | 0,001 | 0,1 | Mn | 0,00005 | 0,1 |
| Cd | 0,00001 | 0,001 | Ni | 0,0005 | 0,1 |
| Co | 0,0005 | 0,1 | Pb | 0,0002 | 0,03 |
| Cr | 0,0005 | 0,05 | Zn | 0,0003 | 5,0 |
1. Основы аналитической химии / Под ред. Ю.А. Золотова. В 2-х т. М.: Высш. шк., 2000.
2. Основы аналитической химии. Практическое руководство / Под ред. Ю.А. Золотова. М.: Высш. шк., 2001.
3. Кунце У., Шведт Г. Основы качественного и количественного анализа / Пер. с нем. М.: Мир, 1997.
4. Пилипенко А.Т., Пятницкий И.В. Аналитическая химия. В 2-х т. М.: Химия, 1990.
5. Юинг Г.Инструментальные методы химического анализа / Пер. с англ. М.: Мир, 1989.
6. Дерффель К. Статистика в аналитической химии / Пер. с нем. М.: Мир, 1994.
7. Кузьмин Н.М., Золотов Ю.А. Концентрирование следов элементов. М.: Наука, 1988.
8. Горелик Д.О., Конопелько Л.А., Панков Э.Д. Экологический мониторинг. В 2 т. СПб.: Крисмас
9. Моисеев Н.Н., Восхождение к разуму. М., «ИЗДАТ», 1993.
10. Венецкий С.И., Рассказы о металлах. М., «Металлургия», 1970.
11. Эйхлер В., Яды в нашей пище. М., «Мир», 1993.
12. Рюдт С, Химия биологически активных природных соединений, М., «Мир», 1978.
13. Штефен Д., Антропогенное загрязнение и здоровье, М., «Мир», 1976. Ревелль П., Ревелль Ч., Среда нашего обитания, книга четвертая, М., «Мир», 1995.
14. Барковский Е.В., Введение в химию биогенных элементов и химический анализ, Минск, «Вышейшая школа», 1997.
15. Назаренко В.Т., Руководство к экологизированному курсу химии, М., «Просвещение», 1995.
16. Николаев Л.А., Химия жизни, М., «Просвещение», 1973.
17. Кукушкин Ю.Н., Химия вокруг нас, М., «Высшая школа», 1992.
18. И.Ю. Пархоменко, В.Л. Таусон, В.И. Меньшиков Термическая атомно-абсорбционная спектроскопия как метод диагностики форм нахождения тяжелых металлов в объектах окружающей среды и минералах
19. Баженова Л.Н., Жернакова З.М., Сулейманова Н.А. Спектрометрические характеристики растворов гумуса. Исследование их комплексообразования с металлами
20. Егорова Л.С., Темерев С.В., Петров Б.И. Определение форм тяжелых металлов в снежном покрове после экстракции тиопирином.
21. С.С.Шацкая, Н.Ф.Глазырина, И.А. Деревягина Изучение поведения токсичных элементов в природных средах методом атомной абсорбции.
22. Рафалюк В.В., Туровская Е.Н., Алемасова А.С. Квантовохимическое моделирование и исследование пиролиза серосодержащих хелатов меди, кадмия свинца атомно-абсорбционным методом.
23. Т.И. Утенкова Разработка методического обеспечения атомного оптического спектрального анализа почв и биологических материалов для экологической экспертизы на токсичные металлы
источник