Нефть и нефтепродукты содержатся в сточных водах нефтехимических и нефтеперерабатывающих производств, а также производств пестицидов, ПАВ и др. [1—4]. Многокомпонентный состав сточных вод нефтехимических производств затрудняет идентификацию отдельных компонентов и методы их обезвреживания. В настоящее время эти стоки классифицируют как мало- и многосернистые. Среднее содержание нефти и нефтепродуктов в сточных водах нефтеперерабатывающих заводов составляет 10 000 мг/л [5].
Пороговая концентрация по привкусу 0,1 мг/л [7]. Пороговая концентрация по запаху разных видов нефтепродуктов: бензин с добавкой нефти 0,00005, дизельное топливо 0,0005, деодорированный керосин 0,82, сырая нефть 0,1— 0,5, мазут 0,22—0,5, нефть очищенная 1,0—2,0 мг/л. В воде, содержащей 0,5 мг/л нефтепродуктов, мясо рыбы приобретает привкус нефти через 1 сут, 0,2 мг/л — через 3 сут, а 0,1 мг/л — через 10 сут [8]. Пороговая концентрация по запаху в мясе рыб 0,1 мг/л . При концентрации 0,25 мг/л мясо форели приобретает привкус через 24 ч, а при 1 мг/л — сразу [9].
Для теплокровных животных при приеме внутрь нефтепродукты малотоксичны. ЛД50 бензина для кроликов 28 350 мг/кг . Нефтяная пленка на поверхности воды пропитывает перья у перелетных птиц, они не могут взлететь и погибают.
Нефть и нефтепродукты относятся к числу трудноокисляемых органических веществ, как на очистных сооружениях канализации, так и в естественных условиях — в водоемах. Неочищенная нефть отличается высокой стабильностью, особенно при низкой температуре воды. В экспериментальных водоемах при низкой температуре воды сохраняет токсичность для водорослей 2 мес. [13]. Нефтепродукты, попавшие в водоем со сточными водами, подвергаются различным изменениям, постепенно опускаются на дно водоема. Бактериальное окисление нефтепродуктов на дне происходит примерно в 10 раз медленнее, чем на поверхности [14]. В водоемах примерно 40% нефти оседает на дне, 40% остается в воде в виде эмульсии и 20% — на поверхности в виде пленки. Нефтяная пленка даже толщиной 0,5 мм на поверхности водоемов затрудняет аэрацию воды, а нефть на дне образует донные отложения; в иле в местах спуска сточных вод обнаружено 3,5—22,0 % нефти [15]. Поэтому при изучении влияния на водоем сточных вод, содержащих нефть, необходимо отбирать не только среднюю пробу, но и отдельные ее фракции (поверхность, глубина примерно 10 см от поверхности, придонные слои и осадок).
Самоочищение водоемов от нефти происходит очень медленно. За 2,7 сут. содержание эмульгированных нефтепродуктов в воде снижалось при 20 °С на 40%, а при 5°С лишь на 15% [16]. В присутствии водной растительности в модельных опытах нефтяная пленка исчезала при ее толщине 0,06 см через 4—6 сут, а при 0,6 см — через 20—22 сут [17]. Следовательно, в водоемах нельзя рассчитывать на самоочищение от нефти. Эти процессы можно использовать лишь при доочистке в биологических прудах.
Нефтепродукты тормозят биологический процесс очистки сточных вод в аэротенках при 50 мг/л [18].
Определение в водных растворах: нефелометрия; весовой метод ;люминесцентный, ИК-спектрометрия, газохроматографический, автоматический метод [19].
Очистка сточных вод: механическая (решетки, отстойники, песколовки, нефтеловушки, песчаные фильтры), физико-химическая (нейтрализация, флотация, окисление кислородом воздуха и озоном, коагуляция), биологическая (аэротенки, аэрируемые пруды на 60 сут пребывания в них сточных вод, биологические фильтры [18, 21—23]. Эффективность очистки сточных вод от нефти на разных типах сооружений составила: нефтеловушки — 99,9%, через песок 50—87%, биофильтры — 47,5%, аэротенки — 53,4% [24]; окисление озоном [25]; биологическая очистка в аэротенках и биологических прудах (при малых концентрациях нефтепродуктов). Нефть и нефтепродукты разлагаются в аэробных условиях микроорганизмами; добавление к сточным водам минеральных солей, хозяйственно-фекальных вод, необходимых для жизнедеятельности микроорганизмов, подача воздуха способствуют более быстрому разложению остатков нефти и нефтепродуктов как на сооружениях биологической очистки в аэротенках, аэрофильтрах и биологических прудах, так и в небольшой степени в водоемах [26]. См. также [27, 28].
- Карелин Я. А., Жуков Д. Д., Денисов М. А. и др. Очистка производственных сточных вод (Опыт Ново-Горьковского нефтеперерабатывающего завода). М., Госстройиздат, 1970. 152 с.
- Хаскин С. А., Карш В. П. — В кн.: Очистка нефтеперерабатывающих сточных вод. М., 1973.
- Wilber Ch. — In: The Biological Aspects of Water Pollution. Springfield, 1969, p. 73.
- Грушко Д. AI., Кожова О. M., Мамонтова Л. М. — Гидробиологический журн., 1978, т. 14, № 2, с. 55.
- Монгайт И. Л., Родзиллер И. Д. — В кн.: Промышленные сточные воды. Вып. 5. М. Медгиз, 1960, с. 7.
- Sittig М. Environmental Sources and Emissions Handbook. Perk Ridge, New Jersey , London, England, 1975. 523 p.
- Гусев А. Г. — Журн. ВХО им. Д. И. Менделеева, 1972, т. 17, № 2, с. 134.
- Гусев А. Г. — В кн.: Производственные сточные воды. Вып. 5. М., Медгиз, 1960, с. 34
- Krishnaswatni S. К., Kupchatiko Е. Е. — J. Water Pollution Control Feder., 1969, v. 41, № 5, part 2, p. R189.
- Мосевич H. А., Гусева H. В., Драгулин M. Г. и dp. — В кн.: Известия ГосВНИОРХ, М., Пищепромиздат, 1952, т. 31, вып. 1, с. 41.
- Миронов О. Г. — Зоологич. журнал, 1969, т. 48, № 7, с. 980.
- Chipman W. A., Galisoff Р. S. Effects of Oil Mixed with Carbonized Sand on Aquatic Animals. Spec. Sci. Rep. Fisher. № 1, U. S. Fish, and Wildlife Service. Wash., 1949. 52 p
- Dickman M. — Artie. Kanad. Field-natur., 1971, v. 85, № 3, p. 249.
- Изъюрова А. И. — Гигиена и санитария, 1950, № 1, с. 9.
- Дадашев X.К., Григорян Э. В., Агамирова С. Н. Сокращение потерь нефтепродуктов с промышленными сточными водами нефтеперерабатывающих заводов. Баку, 1957. 138 с.
- Ломано Л. В., Майер Л. Н., Черепнева В. С. Материалы республиканского научно-технического совещания по изучению, комплексному использованию и охране водных ресурсов. Минск, 1965, с. 41.
- Морозов И. В., Петров Г. /7. — В кн.: Теория и практика биологического самоочищения загрязненных вод. М., Наука, 1972, с. 42.
- Жуков А. И., Демидов Л. Г., Монгайт И. Л. и др. — Канализация промышленных предприятий. Очистка промышленных сточных вод. М., Стройиздат, 1969. 370 с.
- Новиков Ю. В., Сайфутдинов М. М. — Гигиена и санитария, 1977, № 10, с. 60.
- Семенов А. Д., Страдомская А. Г., Павленко Л. Ф. — В кн.: Методы анализа природных и сточных вод. Сер. Проблемы аналитической химии, Т. 5. М., Наука, 1977, с. 220.
- Itieson Pachatn R. — In: Hepple P. (Ed.). Water Pollution by Oil. Proceed, by of Seminar held at Aviemor Invernes — Shiee, Scotland aponsored by the Institute of Water Pollution Control and the Institute of Petroleum, with the Assistance of Eur. Office of WHO, 4—8 May 1970. Amsterdam — London — New York, 1971, p. 143.
- Матвеев AI. C. — Химия и технология топлив и масел, 1962, № 8, с. 24.
- Рубинштейн С. Л., Хаскин С. А. Очистка сточных вод нефтеперерабатывающих заводов, М., ЦНИТЭНефтехим. Сер. «Нефтепереработка и нефтехимия», 1966. 85 с.
- Денисов М. А. Тезисы докладов конференции по методам очистки газовых выбросов и промстоков от вредных веществ. Дзержинск, 1967, с. 12.
- Меренищева Т. Н., Плехоткин В. Ф. Очистка промышленных сточных вод методов озонирования. Обзорная информация. Сер. «Прикладная химия», НИИТЭХим, М., 1974, 21 с.
- Карелин Я. А., Воробьева Г. И. — Химия и технология топлив и масел, 1957, № 10, с. 29.
- Немковский Б. Б., Злобина Г. П., Губанова И. Ф. — Гигиена и санитария, 1962, № 1, с. 61.
- Изъюрова А. И. — Там же, 1958, № 2, с. 72.
- Роговская Ц. И. — В кн.: Биохимический метод очистки производственных сточных вод. М., Стройиздат, 1967, с. 5.
источник
Определение нефти и нефтепродуктов в воде можно осуществлять дифференциальными (газовая, газожидкостная и высокоэффективная жидкостная хроматография, хромато-массспектрометрия) или интегральными (гравиметрия,УФ- и ИК-спектрофотометрия, люминесценция)методами, причем интегральные методы проще и удобнее для проведения наблюдений за состоянием нефтяного загрязнения водоемов и в основном применяются в рутинном анализе.
Однако ни один из перечисленных методов не позволяет получить полную картину качественного состава НП, присутствующих в природных водах. Для исчерпывающей оценки нефтяного загрязнения необходимо применять группу методов. Вместе с тем для практических целей часто бывает вполне достаточно применение какого-либо одного интегрального метода, например ИК-спектрофотометрического или гравиметрического [11].
Гравиметрический метод
Основан на экстракции НП из пробы малополярными растворителями (хлороформ, гексан, четыреххлористый углерод, пентан, петролейный эфир, фреон (хладон) – (1,1,2-трихлор-1,2,2-трифторэтан); очистке экстракта от полярных веществ пропусканием его через колонку с сорбентом (оксид алюминия II степени активности (содержащий 3% H2O), силикагель, флоросил (основной силикат магния), удалении экстрагента путем его выпаривания и взвешивания остатка для определения суммы “нефтепродуктов”.
Обычно для анализа берут 0,1–3 литр воды, подкисляют HCl до рН
В процессе пробоподготовки и хода анализа возможны потери углеводородов, температуры кипения которых менее 100ºС. Непосредственное экстрагирование гексаном приводит к заниженным результатам. Погрешность может доходить до 30%, если исследуемая вода содержит взвешенные частицы [22].
Стандарт ISO 9377-2 не рекомендует применять для экстракции CCl4 из-за его токсичности. В России с конца 1999 г. прекращено производство тетрахлорида углерода как озоноразрушающего вещества.
Основное достоинство гравиметрического метода (одного из немногих “абсолютных” методов аналитической химии) заключается в том, что исключается необходимость использования стандартных образцов такого же качественного и количественного состава, как и исследуемая проба. Также не требуется предварительная градуировка средств измерений. В силу этого метод принят в качестве арбитражного [14].
Предел определения НП в водах при применении хладона составляет 2–4 мг/литр. Арбитражный гравиметрический метод для определения низких концентраций НП требует больших объемов анализируемой воды и растворителей. В связи с этим для повседневной работы по контролю за содержанием НП в питьевой воде и воде водоемов рекомендуется люминесцентно-хроматографический метод [6, 13]. Метод основан на хроматографическом отделении НП от полярных углеводородов и примесей воды не нефтяного происхождения в колонке с активным оксидом алюминия при использовании экстрагентов – хлороформа и гексана и дальнейшем определении выделенных нефтепродуктов люминесцентным методом. Способностью люминесцировать под действием УФ-света обладает лишь часть УВ (ароматические высокомолекулярные, особенно полициклические) и притом по-разному в зависимости от условий возбуждения.
Флуориметрический метод
Флуориметрический метод [14] (по сути мало чем отличающийся от люминесцентно-хроматографического) основан на экстракции нефтепродуктов гексаном, очистке при необходимости экстракта с последующим измерением интенсивности его флуоресценции, возникающей в результате оптического возбуждения. Метод отличается высокой чувствительностью (нижняя граница диапазона измерений 0,005 мг/литр), экспрессностью, малыми объемами анализируемой пробы (100 см3) и отсутствием значимых мешающих влияний липидов. С помощью флуориметрического метода определяются не только нефтепродукты как таковые, но и многие другие органические соединения иного происхождения.
УФ-спектрофотометрический метод
УФ-спектрофотометрический метод для определения НП в ООС применяется достаточно редко, что связано с бесструктурностью спектров поглощения НП. Разработан экспресс-метод определения суммарного содержания нефтепродуктов в воде. Методика определения тяжелых НП основана на их извлечении экстрагентом (гексан, CCl4, хлороформ, толуол) с последующим измерением оптической плотности на спектрофотометре при длинах волн 206; 265; 241 (247); 281(287) нм. Оптимальное время полного извлечения НП – 4 мин, соотношение объемов органической фазы к водной – 1:10, оптимальный интервал рН 4–7 независимо от природы растворителя.
Нижняя граница определяемых концентраций составляет 0,1 мг/литр, длительность анализа – 20мин.
Метод ИК-спектроскопии.
Для мониторинга нефтяных УВ наиболее распространен метод ИК-спектрометрии [7], который позволяет определять сумму алифатических УВ и ПАУ. При этом измеряют содержание как нефтяных УВ антропогенного происхождения, так и продуцируемых морскими организмами [2].
Соответствующие методики анализа основаны на экстракции НП из пробы органическим растворителем (CCl4 или хладон 113), очистке экстракта от полярных соединений методом колоночной хроматографии на оксиде алюминия и последующей регистрации ИК-спектра в области 2700–3200 см-1, обусловленного валентными колебаниями CH3- и CH2-групп алифатических и алициклических соединений и боковых цепей ароматических углеводородов, а также связей CH ароматических соединений.
Метод может быть реализован как в варианте регистрации спектра поглощения в указанной области с помощью традиционного или Фурьеспектрометра, так и в более простом варианте, при котором используется анализатор, измеряющий интегральное поглощение в области 2900–3000 см-1, в которой наблюдаются наиболее интенсивные полосы, соответствующие асимметричным валентным колебаниям групп CH3 и CH2.
ИК-спектроскопия применима для анализа природных вод и промышленных стоков при концентрации нефтяных углеводородов от 0,1 до 50 мг/литр (при использовании кюветы с длиной оптического пути 10 мм), не требует отгонки растворителя и нагрева экстракта, что исключает потерю УВ с низкой температурой кипения.
Преимущество метода ИК-спектроскопии меньшие потери легких фракций, чем при определении НП другими способами [13]. Нижняя граница диапазона измерения – 0,05 мг/литр. Основное достоинство метода – слабая зависимость аналитического сигнала от типа нефтепродукта, составляющего основу загрязнения пробы.
Трудности, возникающие при использовании этого метода, связаны с мешающими влияниями липидов и других полярных соединений при их высоком содержании, при котором оказывается исчерпанной емкость хроматографической колонки, применяемой для очистки экстракта. Основной недостаток метода – его неэкологичность, обусловленная применяемыми высокотоксичными растворителями. В силу указанных причин можно прогнозировать, что уже в ближайшие годы неизбежна замена ИК-спектроскопии другими методами, в первую очередь методом газовой хроматографии.
Методики определения НП в воде, основанные на гравиметрии, УФ-спектрофотометрии, флуориметрии и ИК-спектрометрии, позволяют получить информацию о суммарном содержании неполярных и малополярных УВ нефтяного происхождения. Однако с помощью этих методов нельзя идентифицировать индивидуальные углеводороды НП [13]. Такую задачу решают с помощью газовой хроматографии (ГХ).
источник
Нефтепродукты (НП) относятся к числу наиболее распространенных и опасных веществ, загрязняющих природные воды. Нефть и продукты ее переработки представляют собой сложную, непостоянную смесь предельных и непредельных углеводородов и их различных производных. Понятие «нефтепродукты» в гидрохимии условно ограничивается только углеводородной фракцией (алифатические, ароматические и ациклические), составляющей главную и наиболее характерную часть нефти и продуктов ее переработки. В международной практике содержание в воде нефтепродуктов определяется термином «углеводородный нефтяной индекс» (hydrocarbon oil index).
В связи с неблагоприятным воздействием нефтепродуктов на организм человека и животных, на биоценозы водоемов, контроль за содержанием нефтепродуктов в водах обязателен и регламентируется требованиями ГН 2.1.5.1315-03, ГН 2.1.5.2280-07, СанПиН 2.1.5.980-00, Приказом Росрыболовства от 18.01.2010 №20.
Предельно допустимые концентрации (ПДК) нефтепродуктов в воде водных объектов хозяйственно-питьевого и культурно-бытового водопользования 0,3 мг/дм3, в водах водных объектов рыбохозяйственного значения — 0,05 мг/дм3.
В настоящее время применяют методы определения содержания нефтепродуктов в воде, основанные на различных физических свойствах нефтепродуктов:
- Метод ИК-спектрофотометрии
- Гравиметрический метод
- Флуориметрический метод
- Метод газовой хроматографии.
Метод ИК-спектрофотометрии (ПНД Ф 14.1:2:4.168; МУК 4.1.1013-01, НДП 20.1:2:3.40-08) заключается в выделении эмульгированных и растворенных нефтяных компонентов из воды экстракцией четыреххлористым углеродом, хроматографическом отделении НП от сопутствующих органических соединений других классов на колонке, заполненной оксидом алюминия, и количественном их определении по интенсивности поглощения C-H связей в инфракрасной области спектра. Диапазон измеряемых концентраций: 0,02 – 2,00 мг/дм3. Погрешность методики при Р=0,95 ( ±δ, %): 25 – 50%.
Гравиметрический метод ( ПНД Ф 14.1:2.116-97) основан на извлечении нефтепродуктов из анализируемых вод органическим растворителем, отделении от полярных соединений других классов колоночной хроматографией на оксиде алюминия и количественном определении гравиметрическим методом. Диапазон измеряемых концентраций: 0,30 – 50,0 мг/дм3. Погрешность методики при Р=0,95 ( ±δ, %): 25 – 28% (для природных вод), 10 – 35% (для сточных вод).
Преимуществами этого метода определения НП являются высокая чувствительность и экспрессность анализа.
Методом газовой хроматографии (ГОСТ 31953-2012 ) определяют массовую концентрацию нефтепродуктов в питьевой воде, в том числе расфасованной в емкости, природной (поверхностной и подземной) воде, в том числе воде источников питьевого водоснабжения, а также в сточной воде с массовой концентрацией нефтепродуктов не менее 0,02 мг/дм3.
Метод основан на экстракционном извлечении нефтепродуктов из пробы воды экстрагентом, очистке экстракта от полярных соединений сорбентом, анализе полученного элюата на газовом хроматографе, суммировании площадей хроматографических пиков углеводородов в диапазоне времен удерживания равным и (или) более н-октана ( ) и расчете содержания нефтепродуктов в воде по установленной градуировочной зависимости. Этот метод позволяет определить не только общее содержание нефтепродуктов, но и проводить идентификацию состава нефтепродуктов. Погрешность методики при Р=0,95 ( ±δ, %): 25 – 50%.
В лаборатории АНО «Испытательный Центр «Нортест» измерение массовой концентрации нефтепродуктов в пробах природных, питьевых, сточных вод выполняется флуориметрическим и гравиметрическим методами анализа.
источник
ГОСУДАРСТВЕННОЕ САНИТАРНО-ЭПИДЕМИОЛОГИЧЕСКОЕ НОРМИРОВАНИЕ РОССИЙСКОЙ ФЕДЕРАЦИИ
4.1. МЕТОДЫ КОНТРОЛЯ. ХИМИЧЕСКИЕ ФАКТОРЫ
ОПРЕДЕЛЕНИЕ МАССОВОЙ КОНЦЕНТРАЦИИ
НЕФТЕПРОДУКТОВ В ВОДЕ
1 Разработаны авторским коллективом в составе: Скачков В. Б., Брагина И. В., Ластенко Н. С. (Федеральный центр госсанэпиднадзора Минздрава России), к. х. н. Морозов С. В., Орнацкая Г. Н., Зуева О. А. (Испытательный аналитический центр Новосибирского института органической химии им. Н. Н. Ворожцова Сибирского отделения РАН), при участии к. г. н. Василенко Ю. Г., Петровской И Ф. (Производственно-экологическое предприятие «СИБЭКОПРИБОР», г. Новосибирск).
2. Утверждены и введены в действие Главным государственным санитарным врачом Российской Федерации, Первым заместителем министра здравоохранения Российской Федерации 25 января 2001 года.
санитарный врач Российской
Федерации — Первый заместитель
Дата введения: с момента утверждения
4.1. МЕТОДЫ КОНТРОЛЯ. ХИМИЧЕСКИЕ ФАКТОРЫ
Определение массовой концентрации
нефтепродуктов в воде
Настоящие методические указания устанавливают ИК-фотометрическую методику количественного химического анализа проб питьевых, природных и очищенных сточных вод для определения в них массовой концентрации нефтепродуктов в диапазоне концентраций от 0,02 до 2,00 мг/дм 3 .
Нефтепродукты (НП) — неполярные и малополярные углеводороды (алифатические, ароматические и алициклические), составляющие главную и наиболее характерную часть нефти и продуктов ее переработки ( ГОСТ 17.1.4.01).
Высокие концентрации нефтепродуктов могут оказывать наркотическое действие и вызывать острые отравления. Нефтепродукты, содержащие мало ароматических углеводородов, вызывают наркоз и судороги. Высокое содержание ароматических углеводородов может угрожать хроническими отравлениями.
ПДК нефтепродуктов в воде — не более 0,1 мг/дм 3 .
Методика обеспечивает получение результатов измерений с погрешностью, не превышающей значений, приведенных в табл. 1.
Диапазон измерений, значения характеристики относительной погрешности и ее случайной составляющей при доверительной вероятности Р = 0,95
Характеристика погрешности (границы интервала, в котором погрешность находится с заданной вероятностью), ± d , %
Характеристика случайной составляющей погрешности (среднее квадратическое отклонение случайной составляющей погрешности), s ( d ) ,%
Если массовая концентрация НП в анализируемой пробе воды превышает верхнюю границу диапазона, допускается разбавление элюата (но не более чем в 50 раз) таким образом, чтобы концентрация НП соответствовала регламентированному диапазону; при этом результату количественного химического анализа приписывается значение характеристики погрешности диапазона, в котором произведено измерение.
Измерение массового содержания НП выполняют методом ИК-фотометрии с использованием концентратомера КН-2.
Методика основана на выделении эмульгированных и растворенных нефтяных компонентов из воды экстракцией четыреххлористым углеродом, хроматографическом отделении НП от сопутствующих органических соединений других классов на колонке, заполненной оксидом алюминия, с последующим количественным определением их массовой концентрации по интенсивности поглощения С-Н связей в инфракрасной области спектра на концентратомере КН-2.
Диапазон определяемых концентраций нефтепродуктов (НП) от 0,02 до 2,00 мг/дм 3 .
Мешающее влияние других веществ, присутствующих в пробе воды, устраняется в процессе пробоподготовки.
При выполнении измерений массовой концентрации НП используют следующие средства измерений, вспомогательное оборудование, реактивы и материалы.
Концентратомер КН-2 или другой прибор с аналогичными метрологическими характеристиками. ИШВЖ.004ТУ
Государственный стандартный образец состава раствора НП (углеводородов) в четыреххлористом углероде ГСО 7248
Весы лабораторные ВЛР-200 ГОСТ 24104
Пипетки 2-2-10, 2-2-5, 2-2-1 ГОСТ 29227
Колбы мерные 2-50-2, 2-25-2 ГОСТ 1770
Цилиндры мерные, вместимостью 10, 25, 1000 см 3 ГОСТ 1770
5.2. Вспомогательное оборудование
Шкаф сушильный общелабораторный ГОСТ 13474
Плитка электрическая с закрытой спиралью ГОСТ 14919
Печь муфельная ПМ-8 ТУ 79-337
Стаканы химические, вместимостью 50 см 3 ГОСТ 25336
Стаканчик для взвешивания (бюкс) высокий ГОСТ 25336
Экстрактор либо воронки делительные, вместимостью 0,5- 1,0 дм 3 ГОСТ 25336
Колонки хроматографические с внутренним диаметром 7 мм длиной 200 мм
Штатив для хроматографические колонок
Сито с диаметром отверстий 0,16 мм
Стеклянные палочки длиной 12-15 см
Бутыли из стекла, вместимостью 0,5-1,0 дм 3 , с притертыми пробками для отбора и хранения проб
Четыреххлористый углерод, ГОСТ 20288
Оксид алюминия, для хроматографии, ТУ 6-09-3916
Натрий сернокислый, безводный, ч. (натрия сульфат) ГОСТ 4166
Кислота серная, х. ч. ГОСТ 4204
Кислота азотная ГОСТ 4461
Вода дистиллированная ГОСТ 6709
Стеклоткань или стекловата ГОСТ 10146
6.1. При работе с реактивами соблюдают требования безопасности, установленные для работы с токсичными, едкими и легковоспламеняющимися веществами по ГОСТу 12.1.005.
6.2. При выполнении измерений на концентратомере КН-2 соблюдают правила электробезопасности в соответствии с ГОСТом 12.1.019 и руководством по эксплуатации прибора.
Выполнение измерений может проводить химик-аналитик, владеющий техникой проведения химических работ, изучивший руководство по эксплуатации КН-2.
8.1. Процессы приготовления и подготовки проб к анализу проводят в нормальных условиях согласно ГОСТу 15150 при температуре воздуха (20 ± 5) °С; атмосферном давлении 630-800 мм рт. ст. и влажности воздуха не более 80%.
8.2. Выполнение измерений на концентратомере КН-2 проводят в условиях, рекомендуемых технической документацией к прибору.
Перед выполнением измерений проводят следующие работы: подготовку посуды, четыреххлористого углерода, оксида алюминия, сульфата натрия, очищенной дистиллированной воды, стеклоткани/стекловаты, хроматографических колонок; приготовление градуировочных растворов; подготовку концентратомера КН-2, калибровку/контроль калибровки концентратомера КН-2; отбор проб.
При выполнении измерений массовой концентрации НП необходимо тщательно соблюдать чистоту химической посуды. Для мытья химической посуды разрешается использовать концентрированные серную и азотную кислоты. Запрещается использовать для мытья все виды синтетических моющих средств. Рекомендуется иметь отдельный набор посуды, который используется только для определения НП. Категорически запрещается смазывать шлифы и краны делительных воронок всеми видами смазок!
9.2. Подготовка четыреххлористого углерода
Проверяют чистоту каждой партии в соответствии с руководством по эксплуатации концентратомера КН-2. В кювету заливают чистый четыреххлористый углерод и помещают в прибор. После появления показания нажимают и удерживают в нажатом положении кнопку «О». На табло появится цифровое показание, характеризующее чистоту четыреххлористого углерода. Если это показание лежит в пределах от 10,0 до 20,0 мг/дм 3 , то четыреххлористый углерод пригоден для работы. В противном случае выполняют очистку растворителя путем перегонки в температурном интервале от 76 до 78 °С.
9.3. Подготовка оксида алюминия 2-ой степени активности
Сорбент просеивают через сито и используют мелкую фракцию. Перед употреблением прокаливают в муфельной печи при 600 °С в течение 4 ч, после чего добавляют к прокаленному оксиду алюминия дистиллированную воду (3% масс.) и выдерживают в течение суток при комнатной температуре. При хранении в эксикаторе либо в колбе с притертой пробкой прокаленный оксид алюминия пригоден к использованию в течение 1 месяца.
9.4. Подготовка безводного сульфата натрия
Перед употреблением прокаливают при 400 °С в течение 8 ч. Хранят в эксикаторе.
9.5. Подготовка стеклоткани или стекловаты
Стеклоткань или стекловату выдерживают в разбавленной (1:1) серной или азотной кислоте в течение 12 ч, промывают водопроводной, затем дистиллированной водой и сушат в сушильном шкафу.
Допускается использование ваты медицинской по ГОСТу 5556 (хлопковой, не синтетической!). Перед использованием вату тщательно промывают четыреххлористым углеродом и высушивают при комнатной температуре.
9.6. Подготовка хроматографических колонок
В нижнюю часть вымытой и высушенной колонки помещают комочек стеклоткани или стекловаты. Затем в колонку засыпают 3 г оксида алюминия и вновь помещают слой стеклоткани или стекловаты (0,5 см).
Оксид алюминия используют в колонке однократно.
9.7. Подготовка очищенной дистиллированной воды
Экстрагируют пробу воды из расчета 20 см 3 четыреххлористого углерода на 1 дм 3 воды.
9.8. Приготовление градуировочных растворов
Основной раствор готовят из ГСО 7248-96 состава раствора нефтепродуктов (углеводородов) в четыреххлористом углероде. Для этого в мерную колбу, вместимостью 50 см 3 , с помощью пипетки, вместимостью 1,0 см 3 , помещают 1,0 см 3 ГСО состава НП и доводят объем раствора в колбе до метки четыреххлористым углеродом. Раствор перемешивают и хранят в холодильнике при температуре О-5 °С не более 6 месяцев. Перед использованием раствор выдерживают при комнатной температуре не менее 30 мин. Массовая концентрация полученного раствора 1000 мг/дм 3 . Погрешность приготовления составляет 1,1 % отн.
Рабочий раствор готовят разбавлением основного раствора. Для этого в мерную колбу, вместимостью 50 см 3 , вносят пипеткой 5,0 см 3 основного раствора и доводят объем раствора в колбе до метки четыреххлористым углеродом. Раствор перемешивают и хранят в холодильнике при температуре 0-5 °С не более 3 месяцев. Перед использованием раствор выдерживают при комнатной температуре не менее 30 мин. Массовая концентрация полученного раствора 100 мг/дм 3 . Погрешность приготовления составляет 1,5% отн. Рабочий раствор концентрацией 100 мг/дм 3 используют для калибровки концентратомера КН-2.
Градуировочные растворы готовят для каждого поддиапазона и ближе к нижней границе определяемых содержаний. Градуировочные растворы готовят непосредственно перед использованием путем разбавления рабочего градуировочного раствора. Для этого в мерные колбы, вместимостью 50 см 3 , вносят пипеткой последовательно 10,0; 5,0; 2,5; 1,0 см 3 рабочего градуировочного раствора и доводят объемы растворов в колбах до метки четыреххлористым углеродом. Растворы тщательно перемешивают. Массовая концентрация полученных растворов составляет 20; 10; 5; 2 мг/дм 3 соответственно. Погрешность D (Р = 0,95) градуировочных растворов, обусловленная процедурой приготовления, не превышает 2,5%.
9.9. Подготовка концентратомера КН-2
Подготовку концентратомера КН-2 к работе осуществляют в соответствии с руководством по эксплуатации.
9.10. Калибровка концентратомера КН-2
В кювету заливают чистый четыреххлористый углерод, помещают ее в концентратомер КН-2 и в соответствии с руководством по эксплуатации КН-2 в режиме «калибровка» устанавливают значение «О» шкалы. Используя рабочий раствор массовой концентрации 100 мг/дм 3 , в режиме «калибровка» устанавливают значение «100» шкалы. При отсутствии кюветы в концентратомере в режиме «контроль» считывают цифровое показание «А», которое является контрольным для проверки калибровки.
9.11. Контроль калибровки концентратомера КН-2
Контроль калибровки осуществляют ежедневно.
Контроль калибровки концентратомера КН-2 осуществляют следующим образом: при отсутствии кюветы в концентратомере в режиме «контроль» считывают цифровое показание «а1». Цифровое показание «а1» должно отличаться от цифрового показания «А», полученного по п. 7.10 не более чем на ± 0,5 мг/дм 3 . В противном случае операцию калибровки по п. 7.10 необходимо повторить.
Контроль калибровки в области измеряемых значений массовых концентраций НП проводят с использованием градуировочных растворов, приготовленных по п. 7.8.
9.12.1. Отбор проб воды производится в соответствии с требованиями ГОСТ 17.1.4.01, ГОСТ Р 51592-2000, ГОСТ Р 51593-2000. При отборе должен быть исключен захват пленки НП с поверхности воды. Отобранные пробы помещают в стеклянные сосуды с притертыми пробками, используют полностью и не фильтруют.
Объем отобранной пробы в зависимости от содержания НП в воде должен соответствовать значениям, указанным в табл. 2. Одновременно следует отобрать не менее двух проб из одной точки.
9.12.2. Экстракцию НП из воды производят не позднее 3 часов после отбора пробы. При невозможности проведения экстракции в течение этого срока пробу консервируют добавлением смеси серной кислоты и четыреххлористого углерода из расчета 1 см 3 концентрированной кислоты и 2,0-3,0 см 3 четыреххлористого углерода на 1 дм 3 пробы. При экстракции эти объемы следует учитывать.
Срок хранения консервированных проб воды — 1 месяц с момента отбора.
Пробу анализируемой воды полностью переносят в делительную воронку соответствующей вместимости, приливают разбавленную (1:10) серную кислоту из расчета 1 см 3 на 100 см 3 пробы. Если проба воды была предварительно законсервирована, серную кислоту не добавляют. Сосуд, в котором находилась проба, тщательно ополаскивают 5 см 3 четыреххлористого углерода, затем выливают растворитель в делительную воронку. Прибавляют туда еще 5 см 3 четыреххлористого углерода (с учетом консервации общий объем четыреххлористого углерода в делительной воронке должен быть 10 см 3 ). Выполняют экстракцию, встряхивая делительную воронку не менее 10 мин, затем отстаивают в течение 10 мин. После расслоения фаз нижний слой (экстракт) сливают в стаканчик и подвергают очистке по п. 8.2 или оставляют на хранение. После отделения экстракта измеряют объем пробы в воронке мерным цилиндром.
Экстракт сушат безводным сульфатом натрия (не менее 2 г) в течение 10 мин, добавляя его в стаканчик небольшими порциями при перемешивании содержимого стеклянной палочкой.
В подготовленную по п. 7.6 хроматографическую колонку наливают 3 см 3 четыреххлористого углерода для смачивания. Как только четыреххлористый углерод впитается в оксид алюминия, выливают экстракт. Необходимо следить, чтобы уровень жидкости не опускался ниже слоя оксида алюминия. После прохождения экстракта в колонку вливают дополнительно 3 см 3 четыреххлористого углерода, которым предварительно ополаскивают стенки стаканчика, где проводилась осушка экстракта. Элюат собирают в мерный цилиндр вместимостью 10-25 см 3 . Первые 3 см 3 элюата отбрасывают. Суммарный объем элюата в цилиндре должен составить 10 см 3 (при необходимости доводят до 10 см 3 четыреххлористым углеродом).
Концентратомер КН-2 должен быть предварительно откалиброван. Перед измерением следует провести контроль калибровки прибора.
Элюат заливают в чистую кювету и устанавливают в концентратомер КН-2. Измеряют концентрацию НП в элюате, считывая показания прибора.
В случае, если концентрация НП превышает величину 100 мг/дм 3 , разбавляют элюат четыреххлористым углеродом, затем раствор заливают в кювету, устанавливают в прибор и производят измерение.
Определение массовой концентрации НП в холостой пробе выполняют одновременно с анализом серии проб. Для этого берут 0,5-1,0 дм 3 очищенной (по п. 7.7) дистиллированной воды и обрабатывают ее, как описано в п. 8.
Результаты анализа холостой пробы учитывают при расчете концентрации НП в пробе. Анализ холостой пробы проводят также при использовании новой партии реактивов.
Массовую концентрацию НП (X) в пробе анализируемой воды рассчитывают по формуле:
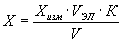 , где (1)
, где (1)
Xизм — содержание НП в элюате, измеренное на приборе, мг/дм 3 ;
V — объем пробы анализируемой воды, см 3 ;
К — коэффициент разбавления, т.е. соотношение объемов мерной колбы и аликвоты элюата (учитывается при его разбавлении по п. 8.3);
V ЭЛ — объем элюата (Уэл = 10 см 3 ).
Из результатов анализа вычитают данные, полученные при анализе холостой пробы по п. 9.
Результат измерения в документах, предусматривающих его использование, представляют в виде:
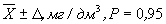 , где (2)
, где (2)
D — значение характеристики погрешности, рассчитанное по формуле:
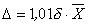 (3)
(3)
( 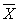 — среднее арифметическое результатов 2-х параллельных определений Х1 и Х2;
— среднее арифметическое результатов 2-х параллельных определений Х1 и Х2; 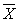 = (Х1 + Х2)/2). Значения d приведены в табл.1.
= (Х1 + Х2)/2). Значения d приведены в табл.1.
14.1. Алгоритм проведения оперативного контроля сходимости
Оперативный контроль сходимости проводят путем сравнения расхождения двух результатов параллельных определений (X1, Х2), полученных при анализе пробы, с нормативом оперативного контроля сходимости — d.
Сходимость результатов параллельных определений признают удовлетворительной, если:
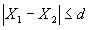 , где (4)
, где (4)
d = 0,01 × dотн × 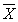
( 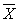 — среднее арифметическое значение двух результатов параллельных определений). Значение dотн приведены в табл. 3.
— среднее арифметическое значение двух результатов параллельных определений). Значение dотн приведены в табл. 3.
При выполнении данного условия по результатам параллельных определений вычисляют результат измерения массовой концентрации НП в рабочей пробе.
При превышении норматива оперативного контроля сходимости эксперимент повторяют. При повторном превышении указанного норматива выясняют причины, приводящие к неудовлетворительным результатам контроля, и устраняют их.
14.2. Алгоритм проведения оперативного контроля воспроизводимости
Образцами для контроля являются две представительные рабочие пробы, отобранные в традиционных точках контроля вод одновременно или непосредственно друг за другом. Пробы анализируют в точном соответствии с прописью методики, получая два результата анализа в разных лабораториях или в одной, причем в этом случае максимально варьируют условия проведения анализа, т.е. используют разные наборы мерной посуды, разные партии реактивов, в работе участвуют два аналитика.
Воспроизводимость результатов измерений рабочих проб признают удовлетворительной,если:
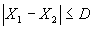 , где (5)
, где (5)
Х1 — средний результат анализа первой рабочей пробы;
Х2 — средний результат анализа второй рабочей пробы в других условиях;
D — норматив оперативного контроля воспроизводимости, причем
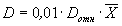 ; (6)
; (6)
( 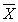 — среднее арифметическое значение результатов анализа первой и второй рабочей пробы). Значения D отн приведены в табл. 3.
— среднее арифметическое значение результатов анализа первой и второй рабочей пробы). Значения D отн приведены в табл. 3.
При превышении норматива оперативного контроля воспроизводимости эксперимент повторяют. При повторном превышении указанного норматива выясняют причины, приводящие к неудовлетворительным результатам контроля, и устраняют их.
Значение нормативов оперативного контроля случайной составляющей относительной погрешности (воспроизводимости и сходимости) при доверительной вероятности Р = 0,95
Диапазон измерений, мг/дм 3
Норматив оперативного контроля воспроизводимости, D отн , % (для двух результатов измерений, m = 2)
Норматив оперативного контроля
(для двух результатов параллельных определений, n = 2)
источник
Нефтепродукты в воде — это опасные вещества, которые негативно влияют на здоровье человека и экологию в целом. Данные примеси портят санитарные условия водоемов и наносят вред народному хозяйству. Большое содержание нефтепродуктов в воде приводит к тому, что водоем становится непригодным для использования.
На загрязнение водоемов нефтяными продуктами влияют два фактора: природный и антропогенный. Последний наносит гораздо больший урон.
- Аварии и разлив нефти при ее добыче
- Аварии при транспортировке и хранении
- Пробоины в нефтепроводах и нефтехранилищах
- Слабая очистка сточных вод на НПЗ
- Заправка водного транспорта
- Выбросы двигателей внутреннего сгорания
Даже малые концентрации нефтепродуктов в воде наносят значительный ущерб здоровью человека. При купании в водоемах с данной примесью есть риск возникновения кожных заболеваний. Употребление питьевой воды, в которой содержатся нефтепродукты, грозит развитием рака внутренних органов, болезней пищеварительной и эндокринной систем, заболеваний полости рта и гортани. Следует провести очистку воды от нефтепродуктов перед ее использованием.
Суммарное количество нефтепродуктов в питьевой воде не должно превышать 0,1 мг на литр; в рыбохозяйственных водоемах – не более 0,05 мг на литр.
Многие предприятия сбрасывают загрязненные сточные воды. Закон регламентирует их место нахождения и запрещает располагаться вблизи водоемов с рыбным хозяйством и питьевых скважинах.
Определить нефтепродукты в воде можно только в лабораторных условиях. Существует несколького способов:
- Гравиметрический – извлечение нефтепродуктов из пробы для анализа при помощи органических растворителей
- Ик-спектрофотометрия – с помощью четыреххлористого углерода путем экстракции выделяются растворенные нефтепродукты
- Флуориметрический – обезвоживание нефтепродуктов и их извлечение с помощью гексана
- Газовая хроматография – экстракционнное извлечение нефтепродуктов с помощью экстрагента
Отбор проб производят только в стеклянную тару. Чтобы провести анализ воды на нефтепродукты, необходимо правильно отобрать материал:
- Слить воду сильным напором в течении 5-10 минут (при отборе пробы из крана)
- Промыть стеклянную тару несколько раз исходной водой (без каких-либо моющих средств)
- Уменьшить напор и отобрать 1,5-2 литра тонкой струей по стенке сосуда
- Закрыть емкость крышкой и незамедлительно доставить в пункт приема проб
Удалить нефтепродукты из воды можно следующими способами:
- Механическим – первичная очистка, которая удаляет 60-65% загрязнений при помощи отстаивания и фильтрации
- Химическим – добавление в сточные воды реагентов, которые разрушают НП
- Физико-химическим – очищение воды от НП посредством коагуляции, флотации и сорбции
- Биологическим – разложение НП с помощью специальных микроорганизмов
В лаборатории «ИОН» вы сможете провести анализ питьевой, природной, талой, морской, технологической воды, а также воды из бассейна и мест общего пользования. Мы работаем более 20-ти лет и занимаемся разработкой новых методов диагностики веществ и материалов. Сотрудники нашей лаборатории – лучшие специалисты в стране, а приборный парк – самый современный, благодаря плодотворному сотрудничеству с крупнейшими разработчиками аналитического оборудования.
Тяжелые металлы – это токсичные и крайне опасные вещества, способные значительно ухудшить здоровье человека и даже привести к гибели. Биогенные элементы – это исключение среди тяжелых металлов, которые необходимы всем живым организмам. Атомный вес тяжелых металлов составляет более 40.
источник
Настоящий документ устанавливает методику выполнения измерений содержаний нефтепродуктов в природных и сточных водах методом колоночной хроматографии с гравиметрическим окончанием при массовых концентрациях нефтепродуктов от 0,30 до 50,0 мг/дм 3
Мешающие влияния, обусловленные присутствием в пробе органических веществ других классов, устраняются в ходе анализа (п. 9).
ʘ Допускается использование данной методики при аварийных ситуациях для определения массовых концентраций нефтепродуктов свыше 50 мг/дм 3 . ʘ
Метод определения массовой концентрации нефтепродуктов основан на извлечении нефтепродуктов из анализируемых вод органическим растворителем, отделении от полярных соединений других классов колоночной хроматографией на оксиде алюминия и количественном определении гравиметрическим методом.
Настоящая методика обеспечивает получение результатов анализа с погрешностью, не превышающей значений, приведенных в таблице 1.
Диапазон измерений, значения показателей точности, повторяемости и воспроизводимости
Показатель точности (границы относительной погрешности при вероятности Р = 0,95), ± d , %
(относительное сред- неквадратическое отклонение повторяемости) s г , %
(относительное среднеквадратическое отклонение воспроизводимости), s R , %
Значения показателя точности методики используют при:
— оформлении результатов анализа, выдаваемых лабораторией;
— оценке деятельности лабораторий на качество проведения испытаний;
— оценке возможности использования результатов анализа при реализации методики в конкретной лаборатории.
При выполнении измерений должны быть применены следующие средства измерений, оборудование и материалы:
3.1. Средства измерений, вспомогательное оборудование
Весы лабораторные, 2 класса точности, ГОСТ 24104
Вентилятор комнатный типа ВН10-УЧ, ГОСТ 7402
Термометр КШ-14/23, ТУ 25-2021.007-88
Стаканчики для взвешивания (бюксы), ГОСТ 25336
Пипетки мерные с делениями 0,1 см 3 4(5)-2-1(2);
Колонка с оксидом алюминия
Бутыли из стекла с притертыми пробками вместимостью 2000 — 3000 см 3 для отбора и хранения проб
Алюминии оксид, ТУ 6-09-3916
Бумага индикаторная универсальная, ТУ 6-09-1181 ʘ
При выполнении измерений массовой концентрации нефтепродуктов в пробах природных и сточных вод соблюдают следующие требования безопасности:
4.1. При выполнении анализов необходимо соблюдать требования техники безопасности при работе с химическими реактивами по ГОСТ 12.1.007.
4.2. Электробезопасность при работе с электроустановками по ГОСТ 12.1.019.
4.3. Организация обучения работающих безопасности труда по ГОСТ 12.0.004.
4.4. Помещение лаборатории должно соответствовать требованиям пожарной безопасности по ГОСТ 12.1.004 и иметь средства пожаротушения по ГОСТ 12.4.009.
Выполнение измерений может производить химик-аналитик, владеющий техникой гравиметрического метода анализа.
При выполнении измерений соблюдают следующие условия:
температура окружающего воздуха (20 ± 5) ℃ ;
атмосферное давление (84 — 106) кПа (630 — 800 мм.рт.ст);
относительная влажность (80 ± 5) %;
частота переменного тока (50 ± 1) Гц;
напряжение в сети (220 ± 10) В.
Отбор проб производится в соответствии с требованиями ГОСТ Р 51592-2000 «Вода. Общие требования к отбору проб» ʘ .
7.1. Пробы воды для параллельных определений отбирают в отдельные стеклянные емкости с притертыми пробками. Пробу для одного определения используют полностью. Если определение нефтепродуктов в день отбора невозможно, то пробы консервируют 2 — 4 см 3 экстрагента (четыреххлористый углерод, хлороформ) на 1 дм 3 воды. Законсервированные пробы могут храниться в течение двух недель.
При определении нефтепродуктов методом колоночной хроматографии с гравиметрическим окончанием объем пробы (при концентрации нефтепродуктов 0,3 — 3,0 мг/дм 3 ) должен составлять не менее 3 — 3,5 дм 3 .
7.2. При отборе проб составляется сопроводительный документ по утвержденной форме, в котором указывается:
— цель анализа, предполагаемые загрязнители;
— должность, фамилия отбирающего пробу, дата.
При подготовке к выполнению измерений проводят следующие работы:
ʘ 8.1. Подготовка оксида алюминия II степени активности ʘ
Реактив перед употреблением прокаливают в муфельной печи при 600 °С в течение 4 часов, дают остыть в эксикаторе и добавляют 3 % (по массе) дистиллированной воды. Хранят в склянке с притертой пробкой.
ʘ 8.2. Подготовка натрия сернокислого безводного ʘ
Перед использованием реактив прокаливают в сушильном шкафу при температуре 105 °С в течение 3 часов.
ʘ 8.3. Подготовка колонки с оксидом алюминия ʘ
Колонка с оксидом алюминия представляет собой стеклянную трубку длиной 10 см и диаметром 0,7 — 1,0 см с оттянутым нижним концом до диаметра 0,1 см. В трубку помещают стеклянную вату слоем 0,5 см, затем 6 г оксида алюминия и снова стеклянную вату. В качестве колонки можно использовать обычную пипетку, градуированную на 10 см 3 . Оксид алюминия в колонке меняют после каждой пробы. Использованный оксид алюминия можно регенерировать промыванием его четыреххлористым углеродом или хлороформом, испарением растворителя и последующим его прокаливанием.
Мешающие влияния, обусловленные присутствием в пробе органических веществ других классов, устраняются в ходе анализа: одни остаются нерастворимыми в гексане, другие (фенолы, нафтеновые кислоты) сорбируются оксидом алюминия.
10.1. Определение при концентрации нефтепродуктов 0,3 — 3,0 мг/дм 3
При выполнении измерений массовой концентрации нефтепродуктов в пробах природных и сточных вод выполняют следующие операции:
3 — 3,5 дм 3 исследуемой пробы воды подкисляют соляной кислотой (плотн. 1,19 г/см 3 ) до рН 3 хлороформа или четыреххлористого углерода, погружают мешалку так, чтобы лопасти её были в воде на 50 мм выше границы слоев воды и растворителя и перемешивают в течение 10 мин.

Затем переносят большую часть водного слоя в другой сосуд такой же вместимости, а оставшийся водный слой и слой хлороформа помещают в делительную воронку вместимостью 500 — 700 см 3 .
Через 15 минут сливают нижний слой хлороформа в коническую колбу (Эрленмейера) вместимостью 500 см 3 , стараясь не захватить при этом ни воды, ни промежуточного слоя эмульсии.
Переливают водный раствор из второго сосуда снова в первый, туда же переносят оставшийся в деятельной воронке водный слой с эмульсией, добавляют вторую порцию хлороформа 150 см 3 и снова перемешивают мешалкой в течение 5 — 7 мин. Снова сливают большую часть водного слоя, остаток переносят в ту же делительную воронку.
Через 15 мин отделяют второй экстракт и присоединяют его к первому, не захватывая при этом водного слоя. Затем небольшим количеством хлороформа (около 50 см 3 ) обмывают стенки сосуда, в котором проба находилась до экстракции, переносят в ту же делительную воронку, взбалтывают, дают некоторое время отстояться и присоединяют слой хлороформа к первым двум экстрактам.
В проведении третьей экстракции обычно нет необходимости.
Экстракцию хлороформом можно также проводить следующим способом: в делительную воронку вместимостью 1 — 2 дм 3 помещают 3 раза по 1 дм 3 исследуемой воды и последовательно взбалтывают с двумя порциями по 20 см 3 хлороформа. Таким образом, на экстракцию из 3 дм 3 анализируемой пробы будет израсходовано 120 см 3 хлороформа. Экстракты соединяют, прибавляют к ним 50 см хлороформного раствора, полученного при ополаскивании сосуда, где хранилась проба (*) .
(*) Склянку, в которой находилась проба, ополаскивают растворителем, который используется для экстракции.
Колбу с экстрактом присоединяют к холодильнику, помещают её в кипящую водяную баню или ставят на горячую закрытую плитку и отгоняют хлороформ до тех пор, пока в колбе не останется 10 — 20 см 3 раствора. Дают колбе остыть и разбирают прибор.
Остатки хлороформа удаляют при комнатной температуре. Предварительно взвешенный бюкс (с крышкой) помещают в вытяжном шкафу на расстоянии 25 — 35 см от обычного комнатного вентилятора, снимают крышку, заполняют бюкс на три четверти полученным экстрактом, включают вентилятор; по мере испарения экстракт подливают в бюкс, пока не перенесут полностью. Колбу из-под экстракта обмывают небольшой порцией хлороформа и переносят в тот же бюкс.
Когда в бюксе останется менее 0,5 см 3 хлороформного раствора, выключают вентилятор и продолжают испарение на воздухе, взвешивая бюкс каждые 2 мин. Перед взвешиванием его закрывают крышкой и вновь снимают крышку для дальнейшего испарения. Когда масса перестанет изменяться, испарение заканчивают.
Разность между массой бюкса с остатком после удаления хлороформа и массой пустого бюкса показывает общее содержание экстрагируемых хлороформом веществ.
Остаток после отгонки хлороформа растворяют в 1 — 2 см 3 предварительно высушенного сульфатом натрия н-гексана или петролейного эфира. Полученный раствор вместе с частицами нерастворившегося остатка, если такие окажутся, переносят в колонку с оксидом алюминия, под которую подставляют чистую сухую колбу. Бюкс несколько раз обмывают маленькими порциями н-гексана, переносят каждую порцию в колонку с оксидом алюминия. Колонку промывают еще несколькими порциями н-гексана (всего 40 — 45 см 3 ), собирая их в ту же колбу. Не следует при этом допускать, чтобы уровень н-гексана в колонке опускался ниже верхней границы слоя оксида алюминия.
Из полученного раствора нефтепродуктов в н-гексане, свободном от полярных соединений, удаляют н-гексан, испаряя его из бюкса при комнатной температуре вентилятором так же, как удаляли раньше хлороформ. Разность между массой бюкса с остатком после удаления н-гексана и массой пустого бюкса показывает содержание нефтепродуктов во взятом для исследования объеме пробы.
10.2. Определение нефтепродуктов в концентрациях выше 3,0 мг/дм 3
Определение проводят так же, как описано в п. 10.1, но только с меньшим объемом исследуемой воды. Берут для анализа 100 — 1000 см 3 воды, соответственно взятому объему воды уменьшают и количество применяемого для экстракции растворителя.
Содержание массовой концентрации нефтепродуктов X (мг/дм 3 ) рассчитывают по формуле:
где m1, — масса бюкса с остатком после удаления гексана, мг,
m2 — масса пустого бюкса, мг;
V — объем пробы, взятой для анализа, см 3 .
За результат анализа Хср принимают среднее арифметическое значение двух параллельных определений Х1 и Х2
для которых выполняется следующее условие:
где r — предел повторяемости, значения которого приведены в таблице 2.
Значения предела повторяемости при вероятности Р = 0,95
Диапазон измерений, мг/дм 3
Предел повторяемости (относительное значение допускаемого расхождения между двумя параллельными результатами измерений), г, %
При невыполнении условия (1) могут быть использованы методы проверки приемлемости результатов параллельных определений и установления окончательного результата согласно раздела 5 ГОСТ Р ИСО 5725-6.
Расхождение между результатами анализа, полученными в двух лабораториях, не должно превышать предела воспроизводимости. При выполнении этого условия приемлемы оба результата анализа, и в качестве окончательного может быть использовано их среднее арифметическое значение. Значения предела воспроизводимости приведены в таблице 3.
Значения предела воспроизводимое при вероятности Р = 0,95
Диапазон измерений, мг/дм 3
Предел воспроизводимости (относительное значение допускаемого расхождения между двумя результатами измерений, полученными в разных лабораториях), R, %
источник
К наиболее распространенным и токсически опасным веществам, которые служат источниками загрязнения природной водной среды, специалисты относят нефтепродукты (НП).
Нефть и её производные являются непостоянными смесями углеводородов предельной и непредельной группы, а также их производных разного вида. Гидрохимия условно трактует понятие «нефтепродукты», ограничиваясь только их углеводородными алифатическими, ароматическими и ациклическими фракциями, которые составляют основную и наиболее распространенную часть нефти и её компонентов, выделяемых в процессе нефтепереработки. Для обозначения содержания нефтепродуктов в воде, в международной практике существует термин Нydrocarbon Оil Index («углеводородный нефтяной индекс»).
Предельная допустимая концентрация (ПДК) в воде нефти и нефтепродуктов для культурно-бытовых и хозяйственно-питьевых объектов водопользования находится на отметке 0,3 миллиграмма на кубический дециметр, а для объектов рыбохозяйственного водопользования – 0,05 миллиграмма на кубический дециметр.
Определение нефтепродуктов, содержащихся в воде, возможно с помощью различных приборов и методов, о которых мы кратко расскажем в этой статье.
На сегодняшний момент существуют четыре основных методики определения концентрации нефти и её производных в воде, которые основаны на разных физических свойствах определяемых нефтепродуктов:
- метод гравиметрии;
- ИК-спектрофотометрия;
- флуориметрический метод;
- методика газовой хроматографии.
Методика применения того или иного способа измерения содержания нефтей и нефтепродуктов в воде, а также нормы ПДК для различных видов нефтепродуктов, регламентируется природоохранными нормативными документами федерального значения (сокращенно – ПНД Ф).
Его применение регулируется ПНД Ф за номером 14.1:2.116-97.
Суть его – извлечение (обезвоживание) нефтепродуктов из предоставленных для анализа проб с помощью органического растворителя, с последующим отделением от полярных соединений с помощью колоночной хроматографии на оксиде алюминия других классов соединений, после чего производится количественное определение содержания вещества в воде.
В исследованиях сточных вод этот способ применяется при концентрациях, диапазон которых составляет от 0,30 до 50,0 миллиграмм на кубический дециметр, что не позволяет определить соответствие воды нормам ПДК на объектах рыбохозяйственного водопользования.
Еще одним существенным недостатком этого способа является длительный период времени, который требуется для проведения измерений. Поэтому его не применяют при текущем технологическом контроле на производстве, а также в других случаях, когда скорость получения результатов имеет первостепенное значение.
К достоинствам этой методики специалисты относят отсутствие стандартных градуировок по образцам, которые характерны для прочих методов анализа.
Погрешность при использовании этого способа при показателе Р равном 0,95 (±δ, %) при анализе природных вод варьируется от 25-ти до 28-ми процентов, а при анализе сточных вод – от 10-ти до 35-ти.
Применение этой методики регламентируется ПНД Ф за номером 14.1:2:4.168, а также методическими указаниями МУК 4.1.1013-01.
Суть этой методики определения содержания нефтепродуктов в воде – выделение растворенных и эмульгированных нефтяных загрязнений путем экстракции их с помощью четыреххлористого углерода, с последующим хроматографическим отделением нефтепродукта от прочих соединений органической группы, на заполненной оксидом алюминия колонке. После этого определение количества НП в воде производится по показателям интенсивности поглощения в инфракрасной области спектра C-H связей.
Инфракрасная спектроскопия на сегодняшний момент является одной из наиболее мощных аналитических методик, и широко применяется в исследованиях как прикладного, так и фундаментального характера. Её применение также возможно для нужд текущего контроля производственного процесса.
Ароматическим углеводородам для возбуждения и последующей регистрации флуоресцентного излучения необходимы различные условия. Специалисты отмечают зависимость спектральных изменений флуоресценции от длины волны, которой обладает возбуждающий свет. Если возбуждение происходит ближней части ультрафиолетового спектра, и уж тем более – в его видимой области, то флуоресценция проявляется только у полиядерных углеводородов.
Так как их доля – достаточно мала, и напрямую зависит от природы исследуемого нефтепродукта, возникает высокая степень зависимости получаемого аналитического сигнала от конкретного вида НП. При воздействии ультрафиолетового излучения люминесцируют только некоторые углеводороды, в основном – высокомолекулярные ароматические из группы полициклических. Причем интенсивность их излучение сильно разнится.
В связи с этим, чтобы получить достоверные результаты, нужно обязательно иметь в наличие стандартный раствор, который содержит те же люминесцирующие компоненты (причем – в таких же относительных пропорциях), что наличествуют в анализируемой пробе. Это чаще всего труднодостижимо, поэтому флуориметрический способ определения содержания в воде нефтепродуктов, который основан на регистрации интенсивности флуоресцентного излучения в видимой части спектра, для массовых анализов является непригодным.
Этот метод можно применять при концентрациях нефтепродуктов в пределах от 0,005 до 50,0 миллиграммов на кубический дециметр.
Погрешность получаемых результатов (при Р равном 0,95, ( ±δ, %)) составляет от 25-ти до 50-ти процентов.
Применение этой методики регулируется ГОСТ-ом за номером 31953-2012.
Эту методику применяют для определения массовой концентрации различных нефтепродуктов как в питьевой (включая расфасованную в емкости), так и в природной (как поверхностной, так и подземной) воде, а также в воде, содержащейся в источниках хозяйственно-питьевого назначения. Эффективен этот способ и при анализе сточной воды. Главное, чтобы массовая концентрация нефтепродуктов была не меньше, чем 0,02 миллиграмма на кубический дециметр.
Суть метода газовой хроматографии заключается в экстракционном извлечении НП из анализируемой пробы воды с помощью экстрагента, последующей его очистке от полярных соединений при помощи сорбента, и заключительном анализе полученного вещества на газовом хроматографе.
Результат получается после суммирования площадей хроматографических пиков выделяемых углеводородов и путем последующего расчета содержания НП в анализируемой пробе воды с помощью заранее установленной градуировочной зависимости.
С помощью газовой хроматографии не только определяют общую концентрацию нефтепродуктов в воде, но и проводят идентификацию их конкретного состава.
Газовая хроматография вообще представляет собой методику, основанную на разделении термостабильных летучих соединений. Таким требованиям соответствует примерно пять процентов от общего числа известных науке органических соединений. Однако именно они занимают 70-80 процентов от общего числа используемых человеком в производстве и быту соединений.
Роль подвижной фазы в этой методике исполняет газ-носитель (обычно инертной группы), который протекает через неподвижную фазу с гораздо большей площадью поверхности. В качестве газа-носителя подвижной фазы применяют:
Чаще всего используется наиболее доступный и недорогой азот.
источник