Непонятное слово «фенилкетонурия» многие мамы впервые слышат в родильном доме, когда на 3 – 5 сутки жизни крохи медицинский работник берёт кровь из пяточки малыша для выявления наследственных заболеваний. Волнующиеся родители часто обеспокоены, нужно ли проводить травматическое исследование абсолютно здоровому малышу, тем более, в семьях обоих родителей никогда не слышали об этих недугах.
К сожалению, иногда результаты неонатального скрининга оказываются положительными, и молодым мамам и папам приходится сталкиваться с лечением редких болезней, в том числе и фенилкетонурии. Давайте разберёмся, насколько опасна эта патология для здоровья малыша, и какие действия нужно предпринять, чтобы снизить до минимума риск негативных последствий.
Фенилкетонурия (ФКУ) – генетическое заболевание, которое связано с нарушением обмена аминокислот, главным образом фенилаланина. Поступление в организм малыша обычной белковой пищи приводит к накоплению в тканях токсических продуктов обмена веществ. Эти соединения необратимо влияют на нервную систему и головной мозг ребёнка, вызывают прогрессирующее слабоумие.
Статистика распространения наследственной болезни отличается в различных странах. Так, в России недуг обнаруживается у одного малыша из 5 – 10 тысяч новорождённых (в зависимости от региона страны). Наиболее безопасной страной в отношении риска развития фенилкетонурии считается Финляндия (1:100000). Турция же занимает первое место по возникновению генетического недуга, один из 2600 новорождённых имеет нарушенный обмен фенилаланина.
Среди больных фенилкетонурией девочки встречаются в 2 раза чаще, чем мальчики. Наследование происходит по аутосомно-рециссивному типу, что означает наличие мутантного гена в обеих семьях родителей. Здоровые носители генетического дефекта могут не догадываться о своей особенности и не иметь никаких внешних проявлений. Ситуация усложняется, когда обладатели мутации вступают в брак и собираются завести детей. Риск появления на свет малыша, больного фенилкетонурией, в таком случае составляет 25%. Половина детей, рождённых в данной семье, останутся бессимптомными носителями наследственного дефекта.
Впервые наследственную природу слабоумия заподозрил норвежский биохимик и психиатр Ивар Асбьёрн Фёллинг. Наблюдая за группой умственно отсталых детей, врач заметил общность клинической картины и обнаружил изменение химического состава мочи больных брата и сестры. Добавив к биологической жидкости раствор хлорного железа, доктор выявил изменение окраски мочи, появление оливково-зелёного оттенка. Наличие изменений анализов у близких родственников дало основание заподозрить наследственные причины заболевания.
Предугадать развитие фенилкетонурии у новорождённого малыша без применения дополнительных исследований невозможно. Кроха ничем не отличается от своих сверстников, выглядит абсолютно здоровым ребёнком. Изменения происходят внутри организма, когда начинается энтеральное вскармливание, в организм поступают белки грудного молока.
Причина патологии кроется в недостатке определённых ферментов, участвующих в превращении незаменимой аминокислоты фенилаланина в тирозин. Таким образом происходит повышение уровня фенилаланина и его производных в крови и спинномозговой жидкости. Продукты обмена веществ оказывают токсическое действие на клетки головного мозга, нарушают жировой обмен и передачу нервных импульсов между нейронами.
В то же время необходимый для нормального функционирования тирозин, не синтезируется в должном количестве. Эта аминокислота является незаменимой и принимает активное участие в образовании гормонов, нейромедиаторов, пигмента меланина.
Если неонатальный скрининг не был проведён в родильном доме, заболевание осталось не распознанным и не были организованы необходимые лечебные мероприятия, то ФКУ проявит себя на первом году жизни крохи. Первые признаки отклонений от нормы родители обнаружат у ребёнка в возрасте 2 – 6 месяцев, когда ранее здоровый карапуз становится вялым и апатичным или наоборот, чрезмерно раздражительным. Приём пищи часто заканчивается срыгиванием, а на коже младенца появляются проявления аллергического дерматита.
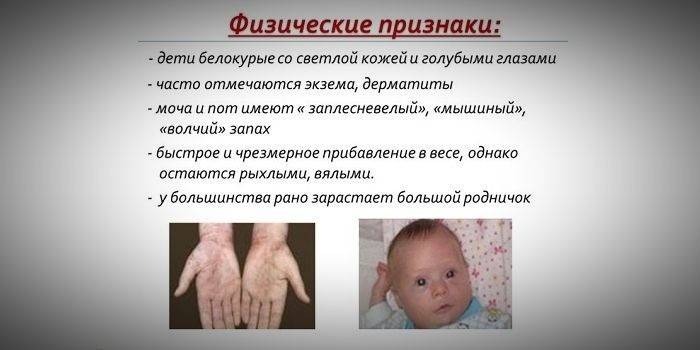
Поскольку нарушение обмена аминокислот приводит к уменьшению образования меланина, в случае развития заболевания кожные покровы, волосы, радужная оболочка глаз теряют пигмент. Малыши с генетическим заболеванием имеют светлые волосы, тонкую бледную кожу, голубые глаза. Указывать на наследственный недуг может характерный запах пота и мочи крохи. Многие врачи характеризуют его как «мышиный», а связан он с выделением продуктов обмена аминокислот с биологическими жидкостями.
Первые признаки изменений со стороны нервной системы проявляются вялостью, снижением тонуса мышц крохи. Нередко возникает дистония, появляются непроизвольные сокращения мышц, навязчивые движения, судороги. Возможно развитие органических патологий головного мозга – микроцефалии и гидроцефалии.
С течением болезни малыш быстро теряет интерес к окружающему миру, становится апатичным. Физическое развитие ребёнка так же страдает – кроха поздно овладевает необходимыми навыками. Задержка в психическом развитии становится очевидной, когда возникают проблемы в формировании речи, отставание в умственном развитии.
Неврологические проблемы у детей старшего возраста выражаются в гиперактивности, появлении стереотипий, навязчивых движений кистей рук, лица, языка, покачиваний. Иногда расстройства психики проявляются в шизофреноподобных состояниях.
Не всегда недуг протекает классически, иногда повышение уровня фенилаланина носит временный характер, а само заболевание не развивается. Непродолжительное увеличение концентрации аминокислоты в крови ребёнка может быть связано с незрелостью ферментных систем малыша, что наиболее характерно для глубоко недоношенных детей. Такие малыши требуют более детального обследования для определения причины заболевания.
Примерно у 2 % малышей, у которых была выявлена фенилкетонурия в родильном доме и назначена соответствующая диета, возникают неврологические проблемы. Эти симптомы могут указывать на развитие у крохи редкой, птерин-зависимой формы ФКУ (тип II, III по классификации). Клинические проявления атипичных форм ФКУ сходно с течением классической формы, но симптомы прогрессируют быстрее, а диетотерапия не оказывает необходимого эффекта.
Огромную роль в развитии птерин-зависимых форм играет дефицит нейромедиаторов, который возникает из-за нарушения обмена аминокислот. Эти вещества необходимы для передачи нервных импульсов между нервными клетками, без которого нормальное функционирование организма невозможно.
Не всегда больные ФКУ дети рождаются от здоровых родителей. Если беременные женщины с наследственной патологией не соблюдают диеты, высока вероятность рождения малыша с синдромом материнской фенилкетонурии. Токсические вещества проникают через плаценту из материнского организма к плоду и нарушают формирование органов и систем ребёнка. На свет появляются дети с множественными врождёнными пороками развития, микроцефалией, умственной и физической отсталостью.
Будущим родителям нужно быть особенно внимательными, если в семьях кого-либо из них имели место случаи рождения больных фенилкетонурией детей. Нельзя исключить возможность появления на свет ребёнка с ФКУ, даже если недуг проявился у дальнего родственника. Коварство заболевания кроется в бессимптомном носительстве повреждённого гена абсолютно здоровыми людьми.
Всем парам, у которых имеются подозрения на наличие наследственных болезней в роду, стоит обратиться к врачу-генетику ещё во время планирования малыша. В медико-генетическом центре (МГЦ) с помощью современных методов диагностики можно выявить носительство мутантного гена и рассчитать риск рождения ребёнка с ФКУ и другими генетическими болезнями.
Перед выпиской из родильного дома новорождённых малышей массово обследуют на наследственные заболевания. Для этого важного исследования медработник набирает кровь из пяточки малыша и наносит капельки биологической жидкости на фильтровальную часть тест-бланка. Каждая капля крови предназначена для выявления одного из 5 заболеваний: фенилкетонурии, муковисцидоза, врождённого гипотиреоза, адреногенитального синдрома, галактоземии.
Этот метод является достоверным в выявлении фенилкетонурии, если малыш получает достаточное количество энтерального питания, грудного молока. Поэтому новорождённым, которые находятся в отделении реанимации, анализ проводят позже. Отличаются и сроки постановки пробы у доношенных и недоношенных детей. Малышам, которые появились в срок, рекомендован набор капиллярной крови на 4-е сутки жизни. Проведение исследование родившимся преждевременно крохам откладывается до 7-х суток.
Анализ следует проводить натощак, что обеспечит более точный результат и повысит диагностическую значимость теста. Бланк с капельками крови малыша и паспортными данными родителей отправляется в лабораторию медико-генетической консультации, где проводится биохимическое исследование.
Чтобы подтвердить заболевание у младенца и установить его причину, малышу придётся пройти многоэтапную процедуру обследования. Повторные биохимические анализы помогут подтвердить повышение уровня аминокислоты в крови и принять решение о необходимости диетотерапии. Проводится исследование уровней фенилаланина и тирозина, активность печёночных ферментов, определение продуктов обмена аминокислот в моче.
Затем осуществляется молекулярно-генетическая диагностика, которая позволяет выявить мутацию в гене РАН, ответственном за развитие фенилкетонурии. С помощью данных методов можно определить и бессимптомное носительство заболевания.
Самым эффективным методом терапии классической формы фенилкетонурии считается правильно организованная диетотерапия. Только исключив из рациона крохи продукты, содержащие большое количество аминокислоты, можно ожидать положительных результатов от лечения.
Фенилкетонурия у детей – непростое заболевание и правильно подобрать рацион для малыша может только врач. Выбирая разрешённые продукты и рассчитывая их объём, доктор учитывает некоторые индивидуальные особенности:
- какая из форм болезни присутствует у малыша;
- степень тяжести недуга, уровень фенилаланина в крови;
- возраст крохи;
- особенности физического развития непоседы;
- физиологическую потребность растущего организма в белке;
- как переносит малыш поступление небольшого количества фенилаланина с пищей.
Поскольку, в большинстве случаев, болезнь определяется ещё в первые недели жизни крохи, становится вопрос о безопасности кормления малыша грудным молоком. На самом деле, грудное вскармливание необходимо для нормально развития ребёнка, становления его иммунной системы. Но применение его оправдано лишь в случаях, когда мать придерживается чётких правил и строго контролирует употребляемый малышом объём съеденной пищи.
Для безопасного кормления лучше использовать свежесцеженное грудное молоко, допустимое количество которого рассчитывает врач исходя из массы тела малыша и его потребности в фенилаланине.
Существуют специально разработанные продукты для рационального питания больных фенилкетонурий малышей. Они изготавливаются на основе гидролизатов белка или смеси аминокислот, которые полностью или частично лишены фенилаланина. Питание лечебными смесями направлено на восполнение организма крохи необходимым белком, достаточное поступление питательных веществ. Эти смеси могут использоваться как заменители грудного молока или применяться, как добавка к основному рациону для старших детей.
С каждым годом малыш растёт, и его потребности в продуктах питания увеличиваются. Приходит время вводить прикорм, пробовать новые блюда. Родителям нужно помнить об опасности фенилаланина для мозга малыша, выбирать только разрешённые врачом продукты.
Все продукты разделяют на группы по содержанию в них фенилаланина:
- красный список.
Рекомендуется полностью исключить продукты этого перечня, поскольку они богаты белками. Это мясо и субпродукты, рыба, яйца, хлеб, гречневая, ячневая крупы, хлопья « Геркулес»;
Допустимо использовать небольшое количество продуктов из этой группы под контролем анализа крови. Возможно введение кукурузной и рисовой крупы, картофеля, молочных продуктов с низким содержанием белка;
Эти продукты составляют основу питания больных фенилкетонурией детей старшего возраста: мука кукурузная и рисовая, большинство овощей, фрукты и ягоды, сладости, сливочное масло.
Применение медикаментов оправдано лишь для лечения птерин-зависимых форм ФКУ, когда диета не оказывает надлежащего эффекта. В таких ситуациях используют препараты L-дофы и 5-гидроокситриптофана.
В остальных случаях возможно использование витаминотерапии, минеральных комплексов, как дополнение к диете. При необходимости назначаются ноотропные препараты, антиконвульсанты. Работа с логопедами, психологами, массаж, физиотерапия так же оказывают положительный результат в лечении недуга.
Течение классической формы заболевания значительно не влияет на продолжительность жизни человека. Но без надлежащего лечения тяжёлые симптомы недуга значительно сказываются на интеллекте и качестве жизни пациента. В случае рациональной диетотерапии и своевременного контроля уровня фенилаланина в крови, дети с ФКУ не отличаются от своих сверстников и хорошо адаптируются в обществе. Атипичные формы недуга нередко приводят к инвалидизации и смерти ребёнка, поскольку лечение редко оказывает хороший результат.
Профилактика заключается в предупреждении близкородственных браков, медико-генетическом консультировании семей из группы риска по развитию ФКУ, проведении массового обследования новорождённых.
Болезнь у малыша – огромный стресс и испытание для молодых родителей, особенно если недуг носит наследственный характер. Но фенилкетонурия – одна из немногих генетических болезней, мероприятия по лечению которой приносят хорошие результаты.
Родителям нужно понимать важность неонатального скрининга и своевременно начатой, правильно подобранной диетотерапии. Стоит внимательно соблюдать все рекомендации лечащего врача и тогда такой страшный диагноз, как фенилкетонурия не станет приговором для ребёнка.
источник
Заболевание, возникновение которого связано с изъянами в генетическом клеточном аппарате, — фенилкетонурия – входит в немногочисленный перечень наследственных болезней, поддающихся лечению. Первооткрывателем этого недуга был врач из Норвегии И.А. Феллинг, позже было выявлено, что за развитие и течение болезни отвечает единственный ген, именуемый геном фенилаланингидроксилазы (длинное плечо 12ой хромосомы, содержащей до 4,5% всего материала ДНК клетки). Наследуемый дефект приводит к частичной или полной деактивации фермента печени фенилаланин-4-гидроксилазы.
Наследственное заболевание фенилкетонурия (ФКУ) приводит к хроническому отравлению организма токсическими веществами, образованных вследствие нарушенного метаболизма аминокислот и процесса гидроксилирования фенилаланина. Постоянная интоксикация вызывает поражение центральной нервной системы (ЦНС), проявлением чего выступает прогрессирующее снижение интеллекта (фенилпировиноградная олигофрения).
Болезнь Феллинга проявляется в избыточном накоплении в организме фенилаланина и продуктов его неправильного обмена. К другим факторам развития фенилкетонурии относится нарушенное транспортирование аминокислот через гематоэнцефалический барьер, низкое количество нейротрансмиттеров (серотонин, гистамин, допамин). При отсутствии своевременного лечения заболевание приводит к умственной отсталости и может стать причиной гибели ребенка.
Причинообразующим фактором возникновения генных нарушений является метаболический блок, который препятствует образованию фенилаланин-4-гидроксилазы (фермент, отвечающий за превращение аминокислоты фенилаланина в тирозин). Протеиногенная аминокислота тирозин является составной частью белков и пигмента меланина, поэтому является необходимым элементом для функционирования всех систем организма, а ее нехватка приводит к ферментопатии.
Следствием подавления образования метаболита, вызванного мутационной инактивацией фермента, является активирование вспомогательных путей обмена фенилаланина. Ароматическая альфа-аминокислота в результате дефектных обменных процессов распадается на токсические производные, которые в нормальных условиях не образуются:
- фенилпировиноградную кислоту (фенилпируват) – жирно-ароматическая альфа-кетокислота, ее образование приводит к миелинизации отростков нейронов и слабоумию;
- фенилмолочную кислоту – продукт, образовавшийся при восстановлении фенилпировиноградной кислоты;
- фенилэтиламин – начальное соединение для биологически активных передатчиков электрохимических импульсов, повышает концентрацию дофамина, адреналина и норадреналина;
- ортофенилацетат – токсичное вещество, вызывающее нарушение обменных процессов жироподобных соединений в головном мозге.
Медицинская статистика свидетельствует о том, что патологически измененный ген присутствует у 2% населения, но при этом он никак не проявляет себя. Генетический дефект передается ребенку от родителей только при наличии заболевания у обоих партнеров, при этом младенец в 50 % случаев становится носителем мутировавшего гена, оставаясь здоровым. Вероятность того, что фенилкетонурия у новорожденных приведет к заболеванию, составляет 25%.
Болезнь Феллинга является генетическим отклонением, наследуемым по аутосомно-рецессивному типу. Такой тип наследования означает, что развитие признаков врожденного заболевания произойдет только при наследовании ребенком по одной дефектной генокопии от обоих родителей, которые являются гетерозиготными носителями видоизмененного гена.
Развитие врожденного заболевания в 99% случаев вызывает мутация гена, ответственного за кодирование фермента, обеспечивающего синтез фенилаланин-4-гидроксилазы (классическая фенилкетонурия). До 1% генетических заболеваний связаны с мутационными изменениями, происходящими в других генах, и вызывающих недостаточность дигидроптеридинредуктазы (ФКУ II типа) или тетрагидробиоптерина (ФКУ III типа).
Классическая форма генетического заболевания у детей в большинстве случаев проявляется во внешне различимых признаках, начиная с 3-9 месяца жизни. Новорожденные, имеющие дефектный ген, выглядят здоровыми, отличительной особенностью бывает специфический хабитус (внешний облик) ребенка. Выраженная симптоматика появляется через 6-12 месяцев после рождения.
ФКУ II типа характеризуется тем, что первые клинические симптомы появляются спустя 1,5 года с момента появления на свет. Признаки заболевания не исчезают после диагностирования генетических отклонений и начала диетотерапии. Этот вид врожденной болезни нередко приводит к летальному исходу на 2-3 году жизни ребенка. Самыми частыми симптомами ФКУ II типа являются:
- выраженные отклонения в умственном развитии;
- гиперрефлексия;
- нарушение двигательных функций всех конечностей;
- синдром бесконтрольных мышечных сокращений.
Клинические признаки мутационных изменений генов III типа сходны с заболеванием, протекающим по II типу. Дефицит тетрагидробиоптерина характеризуется триадой специфических симптомов:
- высокая степень умственной отсталости;
- явно уменьшенный размер черепа по отношению к другим частям тела;
- спастичность мускулатуры (при этом возможна полная обездвиженность конечностей).

В ходе клинических исследований и наблюдений были выдвинуты предположения, что влияние токсичных производных фенилаланинового обмена вызывает снижение интеллектуальных способностей, которое носит прогрессирующий характер и может привести к слабоумию (олигофрении, идиотии). Среди предполагаемых причин необратимых нарушений мозговой деятельности самой обоснованной считается вызванная снижением уровня тирозина нехватка нейромедиаторов, передающих импульсы между нейронами.
Точную причинно-следственную связь между наследственным заболеванием и нарушениями мозга до настоящего времени не выявлено, как и механизм развития вследствие фенилкетонурии таких психических состояний, как эхопраксия, эхолалия, приступов злости и раздражительности. Данные результатов анализов свидетельствую о том, что фенилаланин оказывает прямое токсическое действие на мозг, что тоже может вызывать снижение интеллекта.
Ввиду того, что насыщенность пигментом кожи и волос зависит от уровня тирозина в митохондриях гепатоцитов, а фенилкетонурия приводит к остановке конверсии фенилаланина, пациенты с этим заболеванием имеют фенотипические особенности (рецессивные признаки). Повышенный мышечный тонус становится причиной появления отклонений в телосложении – оно становится диспластическое. К отличительным внешним признакам фенилкетонурии относятся:
- гипопигментация – светлая кожа, бледно-голубые глаза, обесцвеченные волосы;
- синюшность конечностей;
- уменьшенный размер головы;
- специфическое положение тела – при попытках стоять или сидеть ребенок принимает позу «портного» (руки и ноги согнуты в суставах).
При своевременном обнаружении болезнь Феллинга поддается успешному лечению путем корректировки питания, и развитие ребенка происходит в соответствии его возрастной группе. Трудность выявления генной мутации заключается в том, что ранние признаки тяжело обнаружить даже опытному педиатру. Выраженность симптоматики врожденного заболевания усиливается по мере взросления ребенка, потому что употребление белковой пищи способствует развитию нарушений ЦНС.
На протяжении первых дней жизни ребенка признаки патологических отклонений обнаружить трудно – малыш ведет себя естественно, задержки в развитии не наблюдается. Симптомы заболевания впервые начинают проявляться через 2-6 месяцев после рождения. Родителей должно насторожить поведение малыша, которое характеризуется низкой активностью, вялостью, или, наоборот, беспокойством, гипервозбудимостью.
С началом грудного вскармливания в организм новорожденного с молоком начинают поступать белки, что служит катализатором появления первых признаков, однозначно свидетельствующих о том, что заболевание начало прогрессировать. К специфическим клиническим проявлениям болезни относятся:
- постоянная рвота (зачастую принимаемая за врожденное сужение привратника);
- частое срыгивание;
- отсутствие реакции на внешние раздражители;
- мышечная дистония (сниженное напряжение мышц);
- судорожный синдром (судороги эпилептического или неэпилептического характера).
Если манифестация генетической болезни не произошла (или не была замечена) в течение первых 6 месяцев с момента рождения ребенка, то после этого периода уже можно точно определить отставание в психомоторном развитии. Симптомами генетических нарушений, вызванных ферментным дефицитом, у детей, старше полугода, являются:
- снижение активности (вплоть до полной безучастности);
- отсутствие попыток к самостоятельному вставанию, сидению;
- особенный «мышиный» запах кожи (запах плесени возникает вследствие выведения токсических производных фенилаланина через потовые железы и мочу);
- потеря способности к визуальному распознаванию лиц родителей;
- шелушение кожи;
- появление дерматитов, экзем, склеродермии.
Если отклонения в развитии не были выявлены в младенческом возрасте, и соответствующее лечение не проводилось, то заболевание начинает активно прогрессировать и нередко приводит к инвалидности. Отсутствие терапии на раннем этапе болезни вызывает появление следующих симптомов болезни уже в возрасте 1,5 лет:
- микроцефалия (уменьшенные размеры головного мозга);
- прогнатия (смещение верхнего зубного ряда вперед);
- позднее прорезывание зубов;
- гипоплазия эмали (истончение или полное отсутствие зубной эмали);
- задержка речевого развития вплоть до полного отсутствия речи;
- 3, 4 степень олигофрении (задержка психического развития, умственная отсталость);
- врожденные пороки сердца (дефекты в структуре сердечной мышцы, отделах сердца, крупных сосудах);
- расстройства вегетативной системы (акроцианоз, повышенная потливость, артериальная гипотония);
- запоры.
Для проявления мутации с аутосомно-рецессивным характером наследования дефектный ген должен быть унаследован от обоих родителей. Генетические заболевания такого типа встречаются с одинаковой частотой у новорожденных мальчиков и девочек. Патогенез ФКУ предопределяется нарушением обмена фенилаланина, который может протекать в 3 формах. Лечению диетотерапией поддается только классическая фенилкетонурия I типа.
Атипичные формы заболевания нельзя вылечить путем корректировки питания. Эти отклонения вызваны дефицитом тетрагидроптерина, дегидроптеринредуктазы (реже — пирувоилтетрагидроптеринсинтазы, гуанозин-5-трифосфатциклогидролазы и др.). Большинство случаев летальных исходов зафиксировано среди больных редкими вариациями ФКУ, при этом клинические проявления всех форм болезни аналогичны. Риск рождения ребенка с мутировавшим геном фенилаланингидроксилазы возрастает, если его родители являются близкими родственниками (при близкородственных браках).
При подозрении на генетические нарушения диагноз устанавливается на основании совокупности данных, полученных в результате изучения анамнеза болезни – генеалогических сведениях, результатах клинических и медико-генетических исследований. Для своевременного выявления врожденных заболеваний (ФКУ, муковисцидоза, галактоземии и др.) разработана программа обязательного массового обследования в лабораторных условиях всех новорожденных детей (неонатальный скрининг).
Если будущие родители знают о носительстве мутировавшего гена, современная медицина предлагает способы обнаружения дефекта на этапе беременности (дородовая диагностика плода инвазивным методом). Для разделения фенилкетонурии на виды по степени тяжести применяется условная классификация, которая основана на уровне фенилаланина в безфибриногенной жидкости, полученной из плазмы крови:
- Тяжелая фенилкетонурия – 1200 мкмоль/л.
- Средняя – 60-1200 мкмоль/л.
- Легкая (не требует лечения) – 480 мкмоль/л.
Выявление генетических отклонений происходит в несколько этапов. На первом этапе в роддоме у всех младенцев на 3-5 день жизни осуществляется забор периферической крови (из пятки) для исследований. Материал наносится на бумажный бланк и отправляется в биохимическую лабораторию, где происходит его биохимический анализ. На втором этапе скриннинг-теста определяется соответствие концентрации фенилаланина нормальному значению.
Если патологических изменений не обнаружено, диагностика завершается, о чем делается запись в карте ребенка. При наличии отклонений от нормы результаты диагностики направляются врачу-педиатру для обеспечения уточняющего исследования образца крови новорожденного. Здоровье малыша зависит от своевременного и точного выполнения всех мероприятий по выявлению отклонений. Если диагноз подтвердится после повторного скрининг-теста, родители ребенка будут направлены в клинику к детскому генетику для назначения лечения.
Повторная диагностика при обнаружении во время проведения первичного скрининг-теста отклонений от нормы осуществляется путем повторной сдачи анализов. Помимо определения содержания фенилаланина в крови к методам диагностики ФКУ у детей и взрослых относятся:
- проба Феллинга – определение фенилпировиноградной кислоты в моче путем добавления к биоматериалу хлорида железа (происходит окрашивание в сине-зеленый цвет);
- тест Гатри – оценка степени реакции микроорганизмов на продукты обмена или ферменты, содержащиеся в крови пациента;
- хроматография – изучение химических свойств веществ, распределенных между двумя фазами;
- флуориметрия – облучение биоматериала монохроматическим излучением для определения концентрации содержащихся в нем веществ;
- электроэнцефалография – диагностика электрической активности головного мозга;
- магнитно-резонансная томография – возбуждение атомных ядер клеток электромагнитными волнами и измерение их отклика.
В основе терапии фенилкетонурии лежит ограничение потребления продуктов, являющихся источником белков животного и растительного происхождения. Единственным способом успешного лечения является диетотерапия, адекватность которой оценивается по содержанию фенилаланина в сыворотке крови. Предельно допустимый уровень аминокислоты у больных разных возрастных групп составляет:
- у новорожденных и детей до 3 лет – до 242 мкмоль/л;
- у дошкольников – до 360 мкмоль/л;
- у пациентов в возрасте от 7 до 14 лет – до 480 мкмоль/л;
- у подростков – до 600 мкмоль/л.
От того, на каком этапе заболевания произведена коррекция рациона, зависит эффективность диеты. При ранней диагностике врожденной патологии диетотерапия назначается с 8 недели жизни (после этого периода уже начинаются необратимые изменения). Отсутствие своевременных мер приводит к осложнениям и снижению уровня интеллекта на 4 бала за 1 месяц с момента рождения до начала лечения.
Ввиду того, что лечебная диета при фенилкетонурии предполагает полное исключение из рациона животного белка, появляется необходимость применения других источников необходимых аминокислот, а также витаминов группы В, кальций- и фосфорсодержащих минеральных соединений. К продуктам, назначаемым в качестве добавок к безбелковому рациону, относятся:
- белковые гидролизаты (Амиген, Аминазол, Фибриносол);
- не содержащие фенилаланин смеси, насыщенные незаменимыми аминокислотами – Тетрафен, Фенил-фри.
Наряду с лечебными мерами по устранению причины нарушений функционирования организма следует проводить симптоматическое лечение, направленное на устранение дефектов речи и нормализацию координации движений. Комплексная терапия включает физиотерапевтические процедуры, массаж, помощь логопеда, психолога, выполнение гимнастических упражнений. В ряде случае совместно с диетотерапией показан прием антиконвульсантов, ноотропных и сосудистых препаратов.
Фенилкетонурия II и III типа не поддается лечению низкобелковой диетой – уровень фенилаланина в крови остается неизменным при ограничении поступления белка в организм или клиническая симптоматика прогрессирует даже при снижении уровня аминокислоты. Эффективная терапия этих форм заболевания осуществляется с применением:
- тетрагидробиоптерина – фактора пораженного фермента;
- синтетических аналогов тетрагидробиоптерина – эти вещества лучше проникают через гематоэнцефалический барьер;
- препаратов заместительной терапии – не устраняют причину фенилкетонурии, но поддерживают нормальное функционирование организма (Леводопа совместно с Карбидофой, 5-окситриптофан, 5-формилтетрагидрофолат);
- гепатопротекторов – поддерживают функционирование печени;
- противосудорожных средств;
- введения гена фенилаланингидроксилазы в печень – экспериментальный метод.
На первом году жизни ребенка с ФКУ допустимо кормление грудным молоком, но его количество должно быть ограничено. До 6 месяцев допустимым уровнем потребления фенилаланина является 60-90 мг на 1 кг веса малыша (в 100 г молока содержится 5,6 мг фенилаланина). Начиная с 3 месяцев, рацион ребенка следует поэтапно расширять, вводя в него фруктовые соки и пюре.
Детям с 6 месяцев разрешено введение в рацион овощных пюре, каш (из саго), безбелковых киселей. После 7 месяцев можно давать малышу низкобелковые макаронные изделия, с 8 месяцев – хлеб, не содержащий белка. Возраст, до которого следует ограничивать поступление белка в организм больного ребенка, не установлен. Врачи до настоящего времени дискутируют по вопросу целесообразности пожизненной диетотерапии, но сходятся во мнении, что минимум до 18 лет необходимо придерживаться диетического питания.
Фенилкетонурия, диагностированная у женщины, не является поводом отказаться от рождения ребенка. Будущим мамам с ФКУ для предупреждения повреждения плода во время беременности и профилактики возможных осложнений необходимо до планируемого зачатия и во время вынашивания ребенка придерживаться диеты с ограничением фенилаланина (его уровень в крови должен быть до 242 мкмоль/л).
Диета при фенилкетонурии базируется на существенном уменьшении дозы натурального белка в суточном рационе, но организм новорожденного ребенка не может развиваться нормально при отсутствии необходимых микроэлементов. Для восполнения потребности малыша в белке применяются безлактозные аминокислотные смеси, которыми, согласно российскому законодательству, больные должны обеспечиваться бесплатно.
Толерантность грудничка к фенилаланину в течение первого года жизни стремительно изменяется, поэтому необходимо контролировать его концентрацию в крови ребенка и вносить корректировки в диету. Смеси рассчитаны на определенные возрастные группы:
- малышам до года назначаются Афенилак 15, Аналог-СП, PKU-1, PKU-mix, PKU Anamix;
- детям, старше 1 года, назначают обогащенные витаминами и минералами смеси с повышенным содержанием белка – PKU Prima, P-AM Universal, ФКУ-1, ФКУ-2, ХР Максамейд, ХР Максамум.

Одним из основных компонентов пищевой диеты при фенилкетонурии являются малобелковые продукты на основе крахмала. Эти добавки содержат гидролизат казеина, триптофан, тирозин, метионин, азот и обеспечивают суточную потребность организма ребенка в белке, который необходим для нормального развития и роста. Специализированными продуктами, восполняющими нехватку необходимых минералов и аминокислот при их нехватке в рационе питания, являются:
По мере адаптации организма к фенилаланину детям с возраста 5 лет можно постепенно уменьшать ограничения в питании. Расширение рациона происходит путем введения круп, молочных продуктов, мясных изделий. Школьники старших классов имеют уже высокую толерантность к фенилаланину, поэтому в этом возрасте можно продолжить расширение диеты, при этом необходимо отслеживать реакцию на все изменения в питании. Для контроля состояния ребенка применяются следующие способы:
- оценка неврологических показателей, психологического состояния;
- контроль показателей электроэнцефалограммы;
- определение уровня фенилаланина.
В рацион питания пациентов с ФКУ наряду с малобелковыми крахмалистыми продуктами и лечебными смесями входят и продукты натурального происхождения. При составлении меню следует четко рассчитывать количество потребляемого белка и не превышать рекомендованную врачом дозировку. Для исключения токсического влияния на организм разработаны 3 списка продуктов, которые содержат запрещенные (красный), нерекомендованные (оранжевый) и разрешенные (зеленый) позиции.
Фенилкетонурия развивается на фоне отсутствия фермента, превращающего фенилаланин в тирозин, поэтому высокое содержание белка является поводом для отнесения продуктов в запрещенный (красный) список. Позиции из этого перечня следует полностью исключить рациона питания больного ФКУ:
- мясо;
- внутренние органы животных, субпродукты;
- колбасы, сосиски;
- морепродукты (в т.ч. рыба);
- яйца всех птиц;
- кисломолочные продукты;
- орехи;
- плоды бобовых и зерновых культур;
- соевые продукты;
- желатинсодержащие блюда;
- кондитерские изделия;
- аспартам.
Продукты, которые должны дозировано поступать в организм ребенка с диагнозом ФКУ, включены в оранжевый список. Включение в рацион питания позиций из этого перечня допустимо, но в строго ограниченном количестве. Эти продукты хоть и содержат не много белка, но тоже могу повысить уровень фенилаланина, поэтому их употребление не рекомендовано:
- консервированные овощи;
- блюда из картофеля и риса;
- капуста;
- молоко;
- щербет.
Безбелковые продукты разрешены к употреблению больными с диагнозом фенилкетонурия без ограничений. Перед покупкой позиций из зеленого списка необходимо изучить состав, указанный на упаковке, и убедиться, что там не содержится краситель аспартам, содержащий фенилаланин:
- фрукты;
- овощи (за исключением картофеля и капусты);
- ягоды;
- зелень;
- крахмалистые крупы (саго);
- мед, сахар, варенье;
- мучные изделия из кукурузной или рисовой муки;
- масла, жиры (сливочное, растительное, оливковое).
Фенилкетонурия является неизлечимым заболеванием, которое можно перевести в фазу стагнации путем применения диетотерапии и лечебно-профилактических мер. При изменении условий жизни, нарушении режима питания болезнь может опять обостриться, поэтому больным требуется пожизненное наблюдение. Процесс контроля заключается в периодическом определении уровня фенилаланина в крови. Частота сдачи анализов зависит от возраста пациента:
- до 3 месяцев – скрининг крови надо делать еженедельно до получения устойчивых результатов;
- от 3 месяцев до 1 года – 1-2 раза в месяц;
- от 1 до 3 лет – 1 раз за 2 месяца;
- старше 3 лет – ежеквартально.

Кровь для анализов сдается через 3-4 часа после приема пищи. Помимо скрининга развитие ФКУ контролируется путем определения нутритивного статуса, физического, эмоционального развития больного, уровня интеллектуальных способностей и развитости речи. По результатам наблюдений может возникнуть необходимость дополнительной диагностики с привлечением соответствующих специалистов.
источник
Фенилкетонурия (ФКУ). Причины, симптомы, диагностика и лечение фенилкетонурии. Лечение фенилкетонурии: диета и питание.
Фенилкетонурия (ФКУ) – довольно редкое наследственное заболевание, связанное с нарушением обмена аминокислот. Организм больного фенилкетонурией человека не способен расщеплять аминокислоту фенилаланин, которая поступает с белковой пищей. В результате этого, в тканях накапливаются соединения, отравляющие нервную систему и головной мозг в частности. Развивается умственная отсталость (малоумие), вплоть до идиотии. В связи с этим болезнь получила и другое название – фенилпировиноградная олигофрения.
Однако из всех наследственных заболеваний фенилкетонурия, единственное, которое удается полностью нейтрализовать. Сегодня ребенка, рожденного с признаками ФКУ, можно вырастить абсолютно здоровым. Обезопасить мозг малыша удается с помощью специальной диеты, о которой мы расскажем ниже.
В разных странах частота этого заболевания отличается в разы. В России рождается один больной ребенок на 10 000. В некоторых регионах Великобритании этот показатель в два раза выше – 1:5000. Дети на Африканском континенте практически не болеют фенилкетонурией. Среди больных количество девочек почти в два раза превышает количество мальчиков.
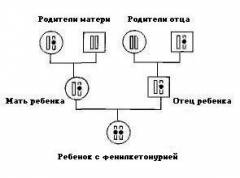 Заболевание наследуется только в том случае, если оба родителя передали ребенку склонность к болезни, и поэтому встречается довольно редко. У двух процентов людей есть измененный ген, который отвечает за развитие болезни. При этом человек остается полностью здоровым. Но когда мужчина и женщина, носители мутировавшего гена, вступают в брак и решают завести детей, то вероятность того, что малыши будут страдать от фенилкетонурии, составляет 25%. А возможность того, что дети будут носителями патологического гена ФКУ, но сами останутся практически здоровыми, составляет 50%.
Заболевание наследуется только в том случае, если оба родителя передали ребенку склонность к болезни, и поэтому встречается довольно редко. У двух процентов людей есть измененный ген, который отвечает за развитие болезни. При этом человек остается полностью здоровым. Но когда мужчина и женщина, носители мутировавшего гена, вступают в брак и решают завести детей, то вероятность того, что малыши будут страдать от фенилкетонурии, составляет 25%. А возможность того, что дети будут носителями патологического гена ФКУ, но сами останутся практически здоровыми, составляет 50%.
Причина возникновения этого заболевания связана с тем, что в печени человека не вырабатывается особый фермент – фенилаланин-4-гидроксилаза. Он отвечает за превращение фенилаланина в тирозин. Последний входит в состав пигмента меланина, ферментов, гормонов и необходим для нормальной работы организма.
При ФКУ фенилаланин, в результате побочных путей обмена, превращается в вещества, которых не должно быть в организме: фенилпировиноградную и фенилмолочную кислоты, фенилэтиламин и ортофенилацетат. Эти соединения накапливаются в крови и оказывают комплексное действие:
- нарушают процессы жирового обмена в мозге
- вызывают дефицит нейромедиаторов, которые передают нервный импульс между клетками нервной системы
- оказывают токсическое действие, отравляя мозг
Это вызывает значительное и необратимое снижение интеллекта. У ребенка быстро развивается умственная отсталость – олигофрения.
 Дети с ФКУ рождаются абсолютно здоровыми. Поэтому, если в течение первых дней жизни выявить заболевание и придерживаться диеты, то удается предотвратить разрушение мозга ребенка. При этом, никакие признаки заболевания не появляются. Малыш развивается и растет, как и его сверстники.
Дети с ФКУ рождаются абсолютно здоровыми. Поэтому, если в течение первых дней жизни выявить заболевание и придерживаться диеты, то удается предотвратить разрушение мозга ребенка. При этом, никакие признаки заболевания не появляются. Малыш развивается и растет, как и его сверстники.
Если же момент упущен, и ребенок употребляет в пищу белковые продукты, богатые фенилаланином, то начинают проявляться симптомы поражения центральной нервной системы. Поначалу изменения у больных фенилкетонурией незначительны. Их трудно заметить даже опытному педиатру. Это слабость и беспокойство. Малыш не улыбается и мало двигается.
К шести месяцам задержка развития становится более заметной. Ребенок слабо реагирует на происходящее, не узнает мать, не пытается сесть и перевернуться. Фенилаланин и его производные выводятся из организма с мочой и потом. Они вызывают специфический «мышиный» или затхлый запах.
Годовалый ребенок не умеет выражать голосом свои эмоции и переживания, имеет невыразительную мимику, не понимает речь родителей.
В возрасте трех лет и старше симптомы фенилкетонурии нарастают. У детей наблюдается повышенная возбудимость, утомляемость, нарушения поведения, психотические расстройства, умственная отсталость. Если не заниматься лечением фенилкетонурии, то состояние больного будет ухудшаться.
 В том случае, если есть подозрение, что один или оба родителя являются носителями гена ФКУ, то определить это можно в федеральных медико-генетических центрах. Для установления этого факта проводится генетическая экспертиза.
В том случае, если есть подозрение, что один или оба родителя являются носителями гена ФКУ, то определить это можно в федеральных медико-генетических центрах. Для установления этого факта проводится генетическая экспертиза.
На сегодняшний день все новорожденные дети массово обследуются на наличие фенилкетонурии. На территории России этот вопрос регламентирует приказ Минздрава РФ №316 от 30.12.1993 г. Процедура получила название неонатальный скрининг и является эффективным способом выявления наиболее распространенных наследственных заболеваний, среди них и ФКУ.
Массовое обследование новорождённых это простой и достоверный метод диагностики. В роддоме у каждого ребенка берут несколько капель периферической крови из пяточки. Это делается натощак, через три часа после кормления. У доношенных детей анализ берут на четвертый день жизни, а у недоношенных на седьмой. У тех новорожденных, которые появились на свет не в родильных домах, важно взять анализ на протяжении первых трех недель.
Кровь наносят на специальный тест-бланк, который потом отправляют в лабораторию для проведения генетического исследования. Там на протяжении суток проводится анализ крови на содержание в ней аминокислоты — фенилаланина. Результаты теста заносятся в обменную карту ребенка в виде штампа: «На ФКУ и ВГ обследован».
В том случае, если в анализе обнаруживают измененный ген, то родителей с ребенком приглашают в медико-генетический центр для обследования. Для того, чтобы подтвердить или опровергнуть диагноз назначаются дополнительные исследования:
- в сухом пятне крови
- в сыворотке крови
- потовый тест
- копрограмма
- ДНК-диагностика
В любом случае родители должны понимать, что при своевременно начатом лечении и соблюдении диеты удается полностью предотвратить развитие болезни.
На сегодняшний день в нашей стране единственным эффективным методом лечения является диетотерапия. Разрабатываются препараты, которые позволят контролировать уровень фенилаланина в крови без соблюдения диеты. В этом направлении есть значительные успехи, но в продаже такие лекарственные средства появятся не раньше чем через 5-7 лет.
Постоянно идет работа над поиском новых средств и методов борьбы с болезнью.
- Перспективным направлением считается использование растительного фермента фенилаланинлиазы, который будет расщеплять излишки фенилаланина в организме.
- Ученые возлагают большие надежды на генотерапию с использованием вирусного фактора, которая позволит вылечить больной ген и полностью избавиться от проблемы.
- Практикуется введение гена фенилаланингидроксилазы прямо в пораженные клетки печени.
Но в нашей стране эти разработки пока не используются. Лечение с помощью диеты – основная помощь больным фенилкетонурией. Ограничить употребление белка необходимо с самого рождения и до половой зрелости. За ростом и развитием ребенка постоянно наблюдают врачи: педиатр и невролог. Специалисты корректируют количество белков, чтобы они соответствовали возрасту и нагрузкам ребенка.
Некоторые формы фенилкетонурии поддаются лечению тетрагидробиоптерином, который является составляющей частью недостающего фермента фенилаланин-4-гидроксилазы. Атипичные формы ФКУ не лечатся с помощью диеты и требуют регулярного приема тетерагидробиоптерина или его заменителей.
 Для того, чтобы нервные клетки ребенка не подвергались токсическому воздействию фенилаланина и его производных, нужно полностью исключить из рациона животные белки. Если это сделать на первых неделях жизни, то мозг останется полностью здоровым. Если же начинать ограничивать белок в более позднем возрасте, то задержку развития удается несколько приостановить. Но вернуть здоровье нервной системе и устранить изменения в нервных клетках уже не удастся.
Для того, чтобы нервные клетки ребенка не подвергались токсическому воздействию фенилаланина и его производных, нужно полностью исключить из рациона животные белки. Если это сделать на первых неделях жизни, то мозг останется полностью здоровым. Если же начинать ограничивать белок в более позднем возрасте, то задержку развития удается несколько приостановить. Но вернуть здоровье нервной системе и устранить изменения в нервных клетках уже не удастся.
Соблюдать диету требуется до 16-18 лет. Это обязательное условие. Желательно контролировать количество животных белков и в дальнейшем.
Если женщина, у которой в детстве были обнаружены признаки ФКУ, планирует забеременеть, то ей обязательно нужно вернуться к диете без фенилаланина. Таких ограничений необходимо придерживаться до зачатия, во время беременности и кормления грудью.
Все необходимые для роста и развития аминокислоты поступают в организм из специализированных лечебных продуктов. Обычно они представляют собой порошок – сухую смесь аминокислот. Их родителям больного малыша выдают бесплатно в медико-генетической консультации.
Грудные дети получают специальные смеси полностью очищенные от лактозы на основе гидролизата молочного белка.
Питательные средства, которые должны заменить детям натуральные белковые продукты, содержат:
- пептиды (расщепленные ферментами молочные белки);
- свободные аминокислоты (тирозин, триптофан, цистин, гистидин и таурин).
В России применяются такие смеси: Афенилак, Аналог-СП, Мдмил-ФКУ-0. Они представляют собой порошки, которые необходимо разводить кипяченой водой или сцеженным грудным молоком, согласно инструкции. В результате получается жидкая смесь или «сметанка». Такой прикорм вводят постепенно, в течение 2-5 дней под наблюдением врача.
Специальными диетическими продуктами для пополнения запасов белка у детей разного возраста также являются: “Берлафен“, “Циморган“, “Минафен“, “Апонти“.
Детей, больных ФКУ, можно кормить грудью. Но при этом кормящей матери нужно придерживаться специальной диеты.
В диете детей дошкольного и школьного возраста полностью исключают из меню белковые продукты. В списке разрешенных продуктов – овощи, фрукты, изделия из крахмала, растительные масла. При составлении дневного меню необходимо строго придерживаться возрастных норм фенилаланина.
| Возраст ребенка | Суточное количество фенилаланина (мк/кг массы тела) |
| Младше 2 мес. | 60 |
| 2-3 мес. | 60-55 |
| 3-6 мес. | 55-45 |
| 6-12 мес. | 45-35 |
| 1-1,5 года | 35-30 |
| 1,5-3 года | 30-25 |
| 3-6 лет | 25-15 |
| Старше 6 лет | 15-10 |
Необходимо помнить, что для растущего организма полноценное питание жизненно необходимо. Так в сутки ребенку требуется 120 мг тирозина на каждый килограмм массы. Поэтому дети и подростки с таким диагнозом должны получать аминокислоты для построения клеток и роста из дополнительных источников. Также обязательно назначают витаминно-минеральный комплекс. Особенно важно, чтобы ребенок получал норму витаминов С, В6 и B1, фолиевой кислоты, железа, кальция и магния. Количество калорий должно быть увеличено на 30% по сравнению с дневной нормой сверстников.
Разделяют три группы натуральных продуктов. В основе классификации лежит количество в них фенилаланина:
- Красный список – продукты, которые необходимо полностью исключить из рациона.
- Оранжевый список – разрешены в небольших количествах под строгим контролем.
- Зеленый список – могут употребляться без ограничений.
| Красный список | Оранжевый список | Зеленый список |
| Все виды мяса | Молочные продукты | Фрукты |
| Колбасные изделия | Рис и кукуруза | Ягоды |
| Все виды рыбы | Овощи (картофель, капуста) | Зелень |
| Морепродукты | Овощные консервы | Овощи |
| Яйца | Рисовая, кукурузная мука | |
| Сыры | Крахмал и саго | |
| Творог | Сахар и варенье | |
| Орехи | Мед | |
| Хлеб и хлебобулочные изделия | Сливочное и растительное масло, топленый жир | |
| Кондитерские изделия | ||
| Крупы и хлопья | ||
| Продукты из сои | ||
| Поп-корн | ||
| Аспартам |
Промышленность выпускает еще две группы продуктов:
- искусственные низкобелковые продукты, специально для диетического питания (хлеб, печенье, макароны)
- готовые пюре для детского питания на основе фруктов.
На основе этих продуктов можно составить полноценное меню, готовить ребенку вкусные, полезные и разнообразные блюда.
Родителям ребенка, больного ФКУ, важно уметь составлять диету и правильно рассчитывать количество фенилаланина. Для этого необходимо иметь под рукой весы, которые дают возможность взвешивать до десятой доли грамма.
 Необходимо контролировать количество фенилаланина. Оно должно находиться в границах 3–4 мг% или 180–240 мкмоль/л.
Необходимо контролировать количество фенилаланина. Оно должно находиться в границах 3–4 мг% или 180–240 мкмоль/л.
Для определения необходимо сделать анализ крови в лаборатории. До трехмесячного возраста это делают еженедельно.
Постепенно врач снижает количество анализов. С тех месяцев до года – один раз в месяц, с года до трех лет – один раз в два месяца. После трех лет частота проверок снижается до одного раза в три месяца. Существует специальная схема, но специалист может изменить ее, исходя из состояния больного.
Делать анализ желательно утром натощак. От качества и регулярности такого контроля и своевременных исправлений в диете зависит сохранение интеллекта.
Новорожденные с диагнозом ФКУ ничем не отличаются от здоровых детей. И если болезнь вовремя выявить и остановить ее развитие, то и в дальнейшем такой ребенок останется абсолютно здоровым.
Как выглядят больные фенилкетонурией? 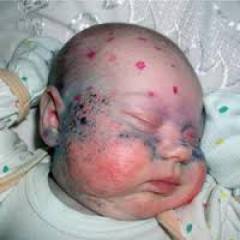
При рождении младенцы, больные фенилкетонурией, ничем не отличаются от остальных детей. Но на втором месяце жизни начинают проявляться изменения:
- посветление волос и радужки глаза из-за недостатка пигмента меланина
- чрезмерная прибавка в весе
- быстро зарастает большой родничок
- суховатая кожа
- шелушение, сыпь и экзема
- частая рвота
- моча и пот с характерным «мышиным» запахом
На втором полугодье дети, не получающие лечение, перестают узнавать мать, не могут фиксировать взгляд на одном предмете, не реагируют на яркие игрушки, не садятся и не переворачиваются, становятся раздражительными. В возрасте 2-3 года отмечаются такие особенности:
- появляются судороги и спазмы
- скованность движений и зажатая «поза портного», что связанно с повышенным напряжением в мышцах
- неадекватное поведение, выкрики, смех
- уменьшение размеров черепа
- деформация ушных раковин
- дрожание пальцев рук
- недержание мочи
- выступающая вперед нижняя челюсть
Внешние признаки болезни выражены незначительно, но при отсутствии диеты развиваются сильные психические отклонения, приводящие к инвалидности.
Для того чтобы обеспечить детей всеми необходимыми веществами в достаточном количестве разработаны специальные смеси на основе незаменимых и заменимых аминокислот. В их состав также входят витамины и все необходимые микроэлементы.
Для детей до одного года рекомендуют:
- Афенилак 13, Афенилак 15 от компании «Нутритек», Россия;
- MIDмил ФКУ 0 (Hero, Испания);
- ХР Аналог («Нутриция», Голландия);
- Фенил Фри 1 («Мид Джонсон» США).
Для детей старше одного года и для взрослых:
- П-АМ 1, П-АМ 2, П-АМ 3;
- Изифен (готовый продукт), а также ХР Максамейд и ХР Максамум с нейтральным и фруктовым вкусами («Нутриция», Голландия).
Эти продукты имеют прекрасные вкусовые качества и хорошо переносятся. Они необходимы детям и взрослым с диагнозом ФКУ в периоды умственных и физических нагрузок. Смеси удобны в применении, питательны и полностью покрывают потребности организма в аминокислотах.
 На сегодняшний день в России для лечения ФКУ используют специальную безфенилаланиновую диету. Для пополнения запасов тирозина и других аминокислот, все больные дети бесплатно получают специальные препараты. Соблюдать диету желательно до 18 лет, хотя ряд врачей утверждает, что лучше делать это на протяжении всей жизни.
На сегодняшний день в России для лечения ФКУ используют специальную безфенилаланиновую диету. Для пополнения запасов тирозина и других аминокислот, все больные дети бесплатно получают специальные препараты. Соблюдать диету желательно до 18 лет, хотя ряд врачей утверждает, что лучше делать это на протяжении всей жизни.
Такой метод диетотерапии является самым дешевым и действенным. На протяжении многих лет он помогает детям с этой болезнью вырасти здоровыми. В своем развитии они ничем не уступают сверстникам. Те, кому в детстве ставили диагноз «фенилкетонурия», учатся в школе, получают высшее образование, заводят семью и рожают здоровых детей.
Подводя итоги, отметим, что фенилкетонурия это тяжелое генетическое заболевание, которое может привести к психической инвалидности. Изменения происходят в нервной системе очень быстро и имеют необратимый характер. Однако можно не допустить развития болезни. Для этого необходима ранняя диагностика и специальная диета.
- ФенилкетонурияI. Классическая и наиболее распространенная форма заболевания, описанная выше в статье. Связана с мутацией гена в 12-й хромосоме, при этом нарушается образование фермента фенилаланин-4-гидроксилазы, который превращает фенилаланин в тирозин.
- ФенилкетонурияII. При этой форме заболевания нарушение происходит в 4-й хромосоме. Нарушается выработка фермента дигидроптеридинредуктазы, который также способствует превращению фенилаланина в тирозин. Заболевание наследуется так же, как и I форма: для того, чтобы родился больной ребенок, необходимо, чтобы носителями гена были оба родителя. Распространенность фенилкетонурии II – 1 случай на 100 000 новорожденных.
- ФенилкетонурияIII. В результате генетических нарушений возникает недостаток фермента 6-пирувоилтетрагидроптеринсинтазы. Наследуется, как и две предыдущие формы заболевания. Распространенность – 1 случай на 300 000 новорожденных.
Критерии установления инвалидности при фенилкетонурии:
- При фенилкетонурии I инвалидность устанавливают только при необратимых нарушениях со стороны центральной нервной системы, которые приводят к неврологическим расстройствам и умственной отсталости.
- При фенилкетонурии II и III типа группу инвалидности устанавливают во всех случаях.
 Специальной профилактики фенилкетонурии не существует. Но некоторые мероприятия помогают правильно оценить риски, вовремя принять необходимые меры:
Специальной профилактики фенилкетонурии не существует. Но некоторые мероприятия помогают правильно оценить риски, вовремя принять необходимые меры:
- Генетическое консультирование. Необходимо людям, планирующим завести ребенка, которые больны или являются носителями неправильного гена, у которых болен хотя бы один близкий родственник или уже родился больной ребенок. Консультирование проводит врач-генетик. Он помогает разобраться, как ген, ответственный за фенилкетонурию, передавался в предыдущих поколениях, каковы риски будущего ребенка. Также генетик помогает с планированием семьи.
- Скрининг новорожденных. Анализ не помогает предотвратить заболевание, но позволяет выявить его максимально рано, пока оно еще не привело к необратимым изменениям в головном мозге.
- Консультации и диета для женщин, страдающих фенилкетонурией. Если вы женщина и страдаете ФКУ, вам следует проконсультироваться с врачом и спросить, когда лучше планировать беременность в вашем случае. Во время беременности нужно соблюдать правильную диету – это помогает предотвратить дефекты развития у ребенка.
Прогноз зависит от формы заболевания и начала лечения, соблюдения диетических рекомендаций, медико-педагогической коррекции.
При фенилкетонурии I, при своевременном начале необходимых мероприятий, прогноз, как правило, благоприятный. Ребенок растет и развивается нормально. Если опоздать с лечением и диетой, то результат будет не таким хорошим.
При фенилкетонурии II и III прогноз серьезнее. Соблюдение диеты не приносит эффекта.
источник