Анализ мочи на фку у новорожденных. Фенилкетонурия — генетические причины заболевания, симптомы, диагностика и лечение
Фенилкетонурия (фенилпировиноградная олигофрения, болезнь Феллинга) – это наследственное заболевание, связанное с нарушением обмена аминокислоты фенилаланина. В результате накопления токсических продуктов из-за неправильного метаболизма развивается отставание в умственном и физическом развитии. Причем появившиеся нарушения в состоянии здоровья необратимы, но своевременная диагностика заболевания может предотвратить все патологические изменения. Лечение заключается в исключении продуктов питания, содержащих фенилаланин. Если такая элиминационная диета применяется практически с рождения, человек вырастает здоровым. Давайте узнаем поподробнее, что же это за заболевание, чем оно проявляется, как диагностируется и лечится.
Впервые описана в 1934 г. доктором Феллингом, откуда и получила свое второе название. Встречается с частотой в среднем 1: 10 000, но в разных странах мира существуют колебания от 1:2600 (в Турции) до 1:100 000 (в Финляндии и Японии), в России от 1:5 000 до 1:10 000.

В основе заболевания лежит генетический дефект – мутация гена 12-й хромосомы (98% всех случаев фенилкетонурии). Это так называемая классическая фенилкетонурия. Ген кодирует количество фермента фенилаланин-4-гидроксилазы. Фермент отвечает за превращение аминокислоты фенилаланина в организме человека в тирозин в клетках печени. Фенилаланин – это аминокислота, которая содержится в белковых продуктах (мясо, рыба, молоко, яйца и другие).
При мутации гена количество фермента снижается, что приводит к накоплению фенилаланина и продуктов промежуточного метаболизма в тканях организма. Организм пытается избавиться от фенилаланина и продуктов его распада, выводя их с мочой.
Подобные нарушения обмена веществ приводят к нарушению строения нервных проводников, снижению образования нейромедиаторов. Все это, наряду с прямым токсическим действием избытка фенилаланина, приводит к развитию умственных нарушений, являющихся основным проявлением заболевания.
Фенилкетонурия наследуется по аутосомно-рецессивному типу, то есть не зависит от пола, и возникает при совпадении двух патологических генов от отца и матери.
Остальные 2% случаев фенилкетонурии связаны с другими генетическими дефектами и зависят от концентрации прочих ферментов (дигидроптеридинредуктазы и др.). Они имеют те же клинические проявления, но не поддаются лечению диетой. Такие варианты относят к атипичному течению заболевания. Среди них принято выделять фенилкетонурию II и III. Генетический дефект при фенилкетонурии II располагается в 4-й хромосоме, при III – в 11-й хромосоме.
Ребенок с фенилкетонурией рождается внешне здоровым, то есть ничем не отличается от других детей. С поступлением пищи в организм начинается попадание белка, а значит и фенилаланина. Последний постепенно накапливается, и обычно к 2 месяцам жизни появляются первые симптомы: вялость или беспокойство, отсутствие интереса к окружающему миру, срыгивания, изменения мышечного тонуса. Иногда срыгивания столь частые и обильные, что возникает подозрение на патологию желудочно-кишечного тракта (пилоростеноз). Ребенок из-за срыгиваний может плохо набирать в весе.
К 4-6 месяцам становится очевидной задержка психического развития. Ребенок не следит за игрушкой, не реагирует на звук, не узнает родителей. Чем дольше продолжается поступление фенилаланина в организм с едой, тем выраженнее нарушения в психической и мыслительной сферах. Развитие речи резко задерживается. Иногда словарный запас может ограничиваться несколькими словами. Если диагноз не будет выставлен и не будет начато лечение, то к 3-4 годам умственные нарушения достигнут степени идиотии (самая тяжелая степень олигофрении).
Особенностью клинического течения фенилкетонурии является необратимость возникших психических и интеллектуальных изменений. То есть при позднем выявлении помочь таким деткам уже нельзя – на всю жизнь они остаются умственно отсталыми.
Физическое развитие также отстает: дети позже начинают держать голову, переворачиваться, сидеть. Когда такие дети начинают ходить, то при этом они широко расставляют ножки, сгибая их одновременно в коленных и тазобедренных суставах. Походка покачивающаяся, мелкими шажками. В положении сидя дети принимают «позу портного» — сгибают и руки, и ноги, поджимая последние под себя. Обычно объем головы меньше, чем в норме. Может быть выраженная микроцефалия: маленькая голова.
Из других неврологических симптомов возможны нарушения мышечного тонуса, судорожные припадки. Эпилептические приступы обычно появляются в возрасте 1,5 лет и приводят к еще большему прогрессированию нарушений интеллекта.
У части больных фенилкетонурией появляются непроизвольные движения в конечностях, дрожание (гиперкинезы). В движениях нет плавности и согласованности, нарушается равновесие.
Кроме ряда психических и интеллектуальных изменений, фенилкетонурию характеризуют следующие симптомы:
- специфический «мышиный» запах (или запах плесени) от ребенка: этот симптом характерен только для фенилкетонурии. Запах появляется в результате выделения продуктов метаболизма фенилаланина (фенилпировиноградной, фенилмолочной, фенилуксусной кислот) через кожу и с мочой;
- кожные проявления: дерматиты, экзема, просто шелушение (возникают по той же причине, что и «мышиный» запах);
- позднее прорезывание зубов: у таких детей первые зубы могут появиться после 18 месяцев, эмаль недоразвита;
- нарушение пигментации: у таких детей обычно голубые глаза, очень светлая кожа и волосы в результате снижения количества меланина (его содержание зависит от метаболизма фенилаланина). Из-за этого у таких детей наблюдается повышенная чувствительность к солнечному свету;
- вегетативные симптомы: пониженное артериальное давление, повышенная потливость, запоры, акроцианоз (синюшность кистей и стоп);
- нередко фенилкетонурия сопровождается врожденными пороками сердца.
Атипичные случаи фенилкетонурии, связанные с нарушением деятельности других ферментов, участвующих в метаболизме фенилаланина, кроме умственных изменений характеризуются развитием мышечной слабости во всех конечностях с одновременным повышением мышечного тонуса, спастическим тетрапарезом. Также при этих формах развивается слюнотечение, приступы повышения температуры.
У взрослых людей, страдающих фенилкетонурией, возможно появление судорожных припадков, нарушений координации, дрожания в конечностях, ухудшения памяти и внимания, возникновение депрессии. Обычно подобные симптомы возникают при несоблюдении элиминационной диеты.
В связи с тем, что фенилкетонурия сопровождается развитием необратимых умственных нарушений, во многих странах мира, в том числе и в России, принято использовать скриниг-методы диагностики. Что это означает? Всем без исключения новорожденным детям в роддоме проводят экспресс-тесты на содержание фенилаланина. Для этого берут капиллярную кровь (из пятки) на 4-5-й день жизни ребенка (у недоношенных на 7-й), наносят на специальный бумажный бланк и отправляют в лабораторию, где по определенным изменениям врач-лаборант делает выводы о содержании фенилаланина в крови. Отрицательный тест говорит об отсутствии фенилкетонурии.
Если тест оказывается положительным, то тогда проводят дополнительные исследования для определения содержания фенилаланина в крови и моче (хроматографию, флюориметрию). Концентрацию фенилаланина в крови и моче регулярно проверяют при проведении лечения, чтобы контролировать эффективность диеты и корригировать ее при необходимости.
Возможно проведение генетического исследования для подтверждения мутации в гене, отвечающем за фенилаланин-4-гидроксилазу. Подобное исследование возможно в качестве пренатальной диагностики, то есть на этапе беременности (берут околоплодные воды путем пункции). Это инвазивное исследование делают по строгим показаниям (например, наличие больного фенилкетонурией ребенка в семье). Выявление генетического дефекта у плода позволяет прервать беременность.
На сегодняшний день самым эффективным и распространенным способом лечения фенилкетонурии является элиминационная диета: диета с исключением продуктов, содержащих фенилаланин. Если ее строго придерживаться в первые годы жизни ребенка, когда развитие нервной системы еще продолжается, то можно вырастить здорового и полноценного человека. Очень важно исключение фенилаланина именно в первый год жизни, когда наиболее активно развивается нервная система. Если элиминационная диета назначается после года, умственные нарушения не излечиваются. Каждый месяц первого года жизни без применения диеты обходится ребенку безвозвратной потерей около 4 баллов IQ. Обычно достаточно придерживаться диеты до 16-18 лет, после этого возраста организм становится менее чувствительным к токсическому действию фенилаланина, и возможно расширение рациона питания. Включение новых продуктов необходимо проводить под контролем содержания фенилаланина в крови. Иногда требуется пожизненное строгое соблюдение диеты. Беременным женщинам и женщинам, планирующим беременность, и при этом больным фенилкетонурией, для рождения здорового ребенка обязательно строгое соблюдение диеты.
Степень строгости диеты зависит от концентрации фенилаланина в крови у ребенка. При его уровне до 2-6 мг% (120-360 мкмоль/л) диета не назначается, выше этого показателя – обязательна.
Суть диеты заключается в исключении белковых продуктов.
Отказ от грудного вскармливания не обязателен, но в этом случае кормящая мать должна строго придерживаться элиминационной диеты, потому что грудное молоко содержит белок (соответственно и фенилаланин). Вопрос о возможности грудного вскармливания решается индивидуально.
 Для пополнения запасов белка назначают специальные смеси, не содержащие фенилаланин – Афенилак, Лофеналак, Нофемикс. После года это Фенилфри, Нофелан, Бигрофен, Тетрафен, МД мил ФКУ-3 и другие. В качестве прикорма назначают овощное и фруктовое пюре, фруктовые кисели, безбелковые каши (рисовая, кукурузная). После 6 месяцев можно применять специальные напитки Лопрофин, Нутриген и другие, кушать макаронные изделия, безбелковый хлеб.
Для пополнения запасов белка назначают специальные смеси, не содержащие фенилаланин – Афенилак, Лофеналак, Нофемикс. После года это Фенилфри, Нофелан, Бигрофен, Тетрафен, МД мил ФКУ-3 и другие. В качестве прикорма назначают овощное и фруктовое пюре, фруктовые кисели, безбелковые каши (рисовая, кукурузная). После 6 месяцев можно применять специальные напитки Лопрофин, Нутриген и другие, кушать макаронные изделия, безбелковый хлеб.
В России обеспечение лечебным питанием детей, больных фенилкетонурией, по закону бесплатное.
Больным фенилкетонурией противопоказаны следующие продукты: мясо, рыба (и морепродукты), орехи, творог, твердый сыр, бобовые, яйца, изделия из пшеничной муки, гречневая и манная крупа, овсяные хлопья.
Во время назначения элиминационной диеты необходим строгий контроль содержания фенилаланина в крови: первые 3 месяца жизни – каждую неделю, от 3-х месяцев до года – минимум раз в месяц, от года до 3-х лет – 1 раз в 2 месяца. Стремятся к содержанию фенилаланина 2-6 мг% у младших детей, после 10 лет – до 10 мг%. Обязательно наблюдение у детского психоневролога.
 Кроме элиминационной диеты периодически назначаются комплексы из витаминов и минералов. Если есть судорожные припадки, необходимо применение антиконвульсантов (Депакин, Клоназепам и другие). Многим из таких детей показан массаж, лечебная физкультура. Возможно использование средств физиотерапии для коррекции мышечного тонуса.
Кроме элиминационной диеты периодически назначаются комплексы из витаминов и минералов. Если есть судорожные припадки, необходимо применение антиконвульсантов (Депакин, Клоназепам и другие). Многим из таких детей показан массаж, лечебная физкультура. Возможно использование средств физиотерапии для коррекции мышечного тонуса.
Атипичные формы фенилкетонурии не поддаются лечению элиминационной диетой. В этом случае показано применение гепатопротекторов, антиконвульсантов, препаратов с Леводопой (для коррекции гиперкинезов), 5-окситриптофана, Тетрагидробиоптерина (ВН 4). Эти формы фенилкетонурии имеют худший прогноз для жизни и тем более интеллектуального развития.
На сегодняшний день разрабатываются новые направления в лечении фенилкетонурии. Среди них стоит отметить следующие:
- использование заместительной терапии фенилаланинлиазой (PAL) – растительным ферментом, расщепляющим фенилаланин до нетоксических соединений;
- генная инженерия (введение искусственно созданного нормального гена, ответственного за фенилаланин-4-гидроксилазу);
- метод «больших нейтральных аминокислот» — уменьшение всасывания фенилаланина из пищи и поступления в головной мозг с помощью специальных препаратов.
Пока эти современные разработки не имеют широкого применения, но некоторые исследования, подтверждающие их эффективность, уже проводятся.
Если женщина, страдающая фенилкетонурией, при планировании беременности и при ее наступлении не придерживается соблюдения диеты, то это отражается на развитии ее ребенка. Потомство таких женщин имеет задержку внутриутробного развития и врожденные пороки развития: пороки сердца, аномалии развития головного мозга, мочевого пузыря, микроцефалию, аномалии лицевого скелета (расщелины).
Чтобы предотвратить патологические изменения у ребенка, таким женщинам необходимо придерживаться элиминационной диеты до зачатия и всю беременность. Дефицит белка восполняется за счет специальных белковых смесей без фенилаланина.
Таким образом, фенилкетонурия – это генетическое нарушение аминокислотного обмена, которое при поздней диагностике приводит к развитию выраженных интеллектуальных нарушений у ребенка. Скрининг этого заболевания в роддомах позволяет диагностировать его в первые недели жизни и вовремя назначить лечение. Основным методом в настоящее время является назначение элиминационной диеты, которая позволяет сохранить интеллект маленькому человечку, а значит, сохранить здоровье, что обеспечит полноценное существование в течение всей жизни.
– наследственное нарушение аминокислотного обмена, обусловленное недостаточностью печеночных ферментов, участвующих в метаболизме фенилаланина до тирозина. Ранними признаками фенилкетонурии служат рвота, вялость или гиперактивность, запах плесени от мочи и кожи, задержка психомоторного развития; типичные поздние признаки включают олигофрению, отставание в физическом развитии, судороги, экзематозные изменения кожи и др. Скрининг новорожденных на фенилкетонурию проводится еще в родильном доме; последующая диагностика включает молекулярно-генетическое тестирование, определение концентрации фенилаланина в крови, биохимический анализ мочи, ЭЭГ, МРТ головного мозга. Лечение фенилкетонурии заключается в соблюдении специальной диеты.

Фенилкетонурия (болезнь Феллинга, фенилпировиноградная олигофрения) – врожденная, генетически обусловленная патология, характеризующаяся нарушением гидроксилирования фенилаланина, накоплением аминокислоты и ее метаболитов в физиологических жидкостях и тканях с последующим тяжелым поражением ЦНС. Фенилкетонурия впервые описана А. Феллингом в 1934 г.; встречается с частотой 1 случай на 10 000 новорожденных. В неонатальном периоде фенилкетонурия не имеет клинических проявлений, однако поступление фенилаланина с пищей вызывает манифестацию заболевания уже в первом полугодии жизни, а в дальнейшем приводит к тяжелым нарушениям развития ребенка. Именно поэтому пресимптоматическое выявление фенилкетонурии у новорожденных является важнейшей задачей неонатологии, педиатрии и генетики.
Фенилкетонурия является заболеванием с аутосомно-рецессивным характером наследования. Это означает, что для развития клинических признаков фенилкетонурии ребенок должен унаследовать по одной дефектной копии гена от обоих родителей, являющихся гетерозиготными носителями мутантного гена.
Чаще всего к развитию фенилкетонурии приводит мутация гена, кодирующего фермент фенилаланин-4-гидроксилазу и расположенного на длинном плече 12 хромосомы (локус12q22-q24.1). Это, так называемая, классическая фенилкетонурия I типа, составляющая 98% всех случаев заболевания. Гиперфенилаланинемия может достигать 30 мг% и выше. При отсутствии лечения данный вариант фенилкетонурии сопровождается глубокой умственной отсталостью.
Кроме классической формы, различают атипичные варианты фенилкетонурии, протекающие с той же клинической симптоматикой, но не поддающиеся коррекции диетотерапией. К ним относятся фенилкетонурия II типа (недостаточность дегидроптеринредуктазы), фенилкетонурия III типа (дефицит тетрагидробиоптерина) и другие, более редкие варианты.
Вероятность рождения ребенка, больного фенилкетонурией, повышается при заключении близкородственных браков.
В основе классической формы фенилкетонурии лежит недостаточность фермента фенилаланин-4-гидроксилазы, участвующего в конверсии фенилаланина в тирозин в митохондриях гепатоцитов. В свою очередь, производный тирозина – тирамин является исходным продуктом для синтеза катехоламинов (адреналина и норадреналина), а дийодтирозин – для образования тироксина. Кроме этого, результатом метаболизма фенилаланина служит образование пигмента меланина.
Наследственная недостаточность фермента фенилалаиин-4-гидроксилазы при фенилкетонурии приводит к нарушению окисления фенилаланина, поступающего с пищей, в результате чего его концентрация в крови (фенилаланинемия) и спинномозговой жидкости значительно возрастает, а уровень тирозина соответственно падает. Избыточное содержание фенилаланина устраняется путем повышенной экскреции с мочой его метаболитов — фенилпировиноградной, фенилмолочной и фенилуксусной кислот.
Нарушение обмена аминокислот сопровождается нарушением миелинизации нервных волокон, снижением образования нейромедиаторов (дофамина, серотонина и др.), запускающими патогенетические механизмы задержки умственного развития и прогредиентное слабоумие.
Новорожденные с фенилкетонурией не имеют клинических признаков заболевания. Обычно манифестация фенилкетонурии у детей происходит в возрасте 2-6 месяцев. С началом кормления в организм ребенка начинает поступать белок грудного молока либо его заменителей, что приводит к развитию первых, неспецифических симптомов – вялости, иногда – беспокойства и гипервозбудимости, срыгивания, мышечной дистонии, судорожного синдрома. Одним из ранних патогномоничных признаков фенилкетонурии служит упорная рвота , которая нередко ошибочно расценивается как проявление пилоростеноза.
Ко второму полугодию становится заметным отставание ребенка в психомоторном развитии. Ребенок становится менее активным, безучастным, перестает узнавать близких, не пытается садиться и вставать на ножки. Аномальный состав мочи и пота обусловливают характерный «мышиный» запах (запах плесени), исходящий от тела. Часто наблюдается шелушение кожи, дерматиты, экзема , склеродермия .
У детей с фенилкетонурией, не получающих лечения, выявляется микроцефалия , прогнатия, позднее (после 1,5 лет) прорезывание зубов , гипоплазия эмали . Отмечается задержка речевого развития , а к 3-4 годам выявляется глубокая олигофрения (идиотия) и практически полное отсутствие речи.
Дети с фенилкетонурией имеют диспластическое телосложение, нередко — врожденные пороки сердца , вегетативные дисфункции (потливость, акроцианоз, артериальную гипотонию), страдают запорами. К фенотипическим особенностям детей, страдающих фенилкетонурией, следует отнести светлую кожу, глаза и волосы. Для ребенка с фенилкетонурией характерны специфическая поза «портного» (согнутые в суставах верхние и нижние конечности), тремор рук, шаткая, семенящая походка, гиперкинезы .
Клинические проявления фенилкетонурии II типа характеризуются тяжелой степенью умственной отсталости, повышенной возбудимостью, судорогами, спастическим тетрапарезом, сухожильной гиперрефлексией. Прогрессирование заболевание может приводить к гибели ребенка в возрасте 2-З лет.
При фенилкетонури III типа развивается триада признаков: микроцефалия, олигофрения, спастический тетрапарез.
В настоящее время диагностика фенилкетонурии (а также галактоземии , врожденного гипотиреоза , адрено-генитального синдрома и муковисцидоза) входит в программу неонатального скрининга, осуществляемого всем новорожденным.
Скрининг-тест проводится на 3-5 день жизни доношенного и 7 день жизни недоношенного ребенка путем забора образца капиллярной крови на специальный бумажный бланк. При обнаружении гиперфенилаланемии более 2,2 мг% ребенка направляют к детскому генетику для повторного обследования.
Для подтверждения диагноза фенилкетонурии проверяется концентрация фенилаланина и тирозина в крови, определяют активность печеночных ферментов (фенилаланингидроксилазы), выполняется биохимическое исследование мочи (определение кетоновых кислот), метаболитов катехоламинов в моче и др. Дополнительно проводится ЭЭГ и МРТ головного мозга, осмотр ребенка детским неврологом.
Генетический дефект при фенилкетонурии может быть обнаружен еще на этапе беременности в ходе инвазивной пренатальной диагностики плода (хорионбиопсии , амниоцентеза , кордоцентеза).
Дифференциальный диагноз фенилкетонурии проводят с внутричерепной родовой травмой новорожденных, внутриутробными инфекциями , другими нарушениями обмена аминокислот.
Основополагающим фактором в лечении фенилкетонурии является соблюдение диеты, ограничивающей поступление белка в организм. Лечение рекомендуется начинать при концентрации фенилаланина >6 мг%. Для грудных детей разработаны специальные смеси — Афенилак, Лофенилак; для детей старше 1 года – Тетрафен, Фенил-фри; старше 8 лет — Максамум-ХР и др. Основу диеты составляют низкобелковые продукты — фрукты, овощи, соки, белковые гидролизаты и аминокислотные смеси. Расширение диеты возможно после 18 лет в связи с возрастанием толерантности к фенилаланину. В соответствии с российским законодательством обеспечение лиц, страдающих фенилкетонурией, лечебным питанием, должна осуществляться бесплатно.
Больным назначается прием минеральных соединений, витаминов группы В и др.; по показаниям — ноотропные средства, антиконвульсанты. В комплексной терапии фенилкетонурии широко используется общий массаж , ЛФК , иглорефлексотерапия .
Дети, страдающие фенилкетонурией, находятся под наблюдением участкового педиатра и психоневролога; нередко нуждаются в помощи логопеда и дефектолога. Необходим тщательный мониторинг нервно-психического статуса детей, контроль уровня фенилаланина в крови и показателей электроэнцефалограммы.
Атипичные формы фенилкетонурии, не поддающиеся лечению диетой, требуют назначения гепатопротекторов, противосудорожных средств, заместительной терапии леводопой, 5-гидрокситриптофаном.
Проведения массового скрининга на фенилкетонурию в неонатальном периоде позволяет организовать раннюю диетотерапию и предотвратить тяжелые церебральные повреждения, нарушения функции печени. При раннем назначении элиминационной диеты при классической фенилкетонурии прогноз развития детей хороший. При поздно начатом лечении прогноз в отношении умственного развития неблагоприятный.
Профилактика осложнений фенилкетонурии заключается в проведении массового скрининга новорожденных, раннего назначения и длительного соблюдения диетического питания.
С целью оценки риска рождения ребенка с фенилкетонурией предварительное генетическое консультирование должны пройти супружеские пары, уже имеющие больного ребенка, состоящие в кровнородственном браке, имеющие родственников с данным заболеванием. Женщины с фенилкетонурией, планирующие беременность, должны соблюдать строгую диету до зачатия и во время беременности для исключения повышения уровня фенилаланина и его метаболитов и нарушения развития генетически здорового плода. Риск рождения ребенка с фенилкетонурией у родителей-носителей дефектного гена, составляет 1:4.
Фенилкетонурия представляет собой достаточно тяжелое наследственное заболевание, основная тяжесть проявлений которого сосредоточена, прежде всего, на нервной системе. Фенилкетонурия, симптомы которой чаще всего встречаются среди девочек, возникает по причине нарушения обмена аминокислот, учитывая же поражение при этом ЦНС, ее проявления сводятся к нарушению умственного развития.
В классической своей форме, которая актуальна для большинства случаев, фенилкетонурия, которую также принято определять как фенилпировиноградную олигофрению, связана с резкостью снижения активности печеночного фермента. Им в частности является фениланин-4-гидроксилаз. Нормальное состояние организма предусматривает катализацию им превращения в тирозин фенилаланина. 1% случаев отмечается возникновением атипичной формы фенилкетонурии, которая возникает по причине мутаций в другого типа генах, которые ответственны за процесс кодирования ферментов. Наследование заболевания происходит в соответствии с аутосомно-рецессивной схемой.
Что касается процессов, происходящих при рассматриваемом заболевании, то они заключаются в следующем. Возникновение характерного метаболического блока провоцирует активацию побочных путей по обмену фенилаланина, что приводит к накоплению токсичных производных от его действия. К таким производным в частности относятся фенилмолочная и фенилпировиноградные кислоты, практически не образующиеся при нормальном состоянии организма. Они же влияют на ЦНС, провоцируя нарушения белкового обмена, обмена липопротеидов и обмена гликопротеидов. Одновременно с этим возникают расстройства в транспорте аминокислот, нарушения в обмене серотонина и катехоламинов, актуальность приобретают также и перинатальные факторы.
Помимо этого начинают также образовываться и в норме практически отсутствующие ортофенилацетат и фенилэтиламин. Их избыток провоцирует нарушения в метаболизме липидов, происходящем в головном мозге. Как предполагается, именно это становится причиной прогрессирующего состояния снижения у больных данным заболеванием интеллекта, что может достичь даже идиотии. В целом же окончательно механизм, в соответствии с действием которого происходит нарушение в развитии функций мозга в случае с актуальным для организма заболеванием фенилкетонурией, не ясен.
Здесь, прежде всего, важно отметить, что первые недели жизни ребенка не позволяют внешне определить данного заболевания. Первые его признаки проявляются через два-шесть месяцев с момента рождения малыша. Он становится вялым, наблюдается отсутствие заинтересованности в отношении условий, его окружающих и мира в целом. Также ребенок становится беспокойным, нарушениям подвергается мышечный тонус. Появляется рвота, судороги, тяжелые кожные . Под экземами в частности подразумевается острая или хроническая форма воспалительного и незаразного заболевания, при котором образуется сыпь. Природа заболевания аллергическая, дополнительными проявлениями симптоматики выступает чувство жжения и выраженный зуд кожи. Актуальна и склонность к рецидивам, то есть к повторному возникновению симптоматики после относительного временного его затишья.
Шестой месяц позволяет определить отставание в развитии у ребенка. Одновременно с этим теряется способность к фокусированию взгляда на отдельных предметах, малыш перестает узнавать родителей. Отсутствует реакция на цветные/яркие игрушки. Важно оперативно приступить к лечению, в противном случае отсталость развития будет постепенно подвергаться лишь прогрессированию в актуальных для нее процессах.
Физическое развитие больных младенцев на физическом уровне отмечается меньшими нарушениями, нежели на психическом. В обхвате голова может быть несколько меньших размеров, чем это предусмотрено для показателей нормы. Зубки прорезываются позже, позже ребенок начинает сидеть и ходить. Принятие положения стоя у таких малышей сопряжено с широким расставлением для этого ног, а также со сгибанием их в коленях и в тазобедренных суставах, плечи и голова при этом опущены. Что касается особенностей ходьбы у больных детей, то она характеризуется покачиваниями, шажки небольшие. Сидят дети, поджав под себя ножки, что обуславливается значительным мышечным тонусом, который они испытывают.
Отличаются детки и характерной внешностью со светлыми волосами, кожа у них абсолютно белая, без пигментации, глаза светлые. Учитывая излишнюю белизну кожи, она нередко покрывается у детей сыпью, что объясняется ее особой чувствительностью в отношении воздействия ультрафиолетового излучения.
В числе основных проявлений, свойственных фенилкетонурии, можно также выделить характерный «мышиный» запах, в некоторых случаях возможны , которые, однако, с возрастом исчезают. Выраженными проявлениями выступают синюшность конечностей, дермографизм (изменение в окраске кожи местного типа, происходящее при механическом воздействии на нее), потливость. Чаще среди больных фенилкетонурией отмечается, помимо перечисленных симптомов, наличие , частых запоров, тремор (т.е. дрожание), потеря равновесия и расстройство в виде нарушения координации движений.
Важным, как мы уже отметили, является раннее диагностирование заболевания, что позволит избежать его развития и привести к ряду необратимых и тяжелых последствий. По этой причине в родильных домах к 4-5 дням жизни (для новорожденных доношенных) берется для анализа кровь. У недоношенных детей на предмет фенилкетонурии (ФКУ) кровь берется на 7 день.
Процедура предусматривает взятие капиллярной крови по прошествии часа с момента кормления, ею в частности пропитывается специальный бланк. Концентрация, указывающая на отметку свыше 2,2% фенилаланина в крови малыша, требует направления его с родителями для осмотра в медико-генетический центр. Там же проводится дообследование и, собственно, уточнение диагноза.
Фенилкетонурия может быть спровоцирована следующими факторами:
- Близкородственные браки, при которых, помимо иных патологий, повышается вероятность рождения ребенка с этим заболеванием;
- Мутация гена (т.е. его изменение), произошедшая по тем или иным причинам в области локализации 12 хромосомы.
Сам процесс наследования гена ФКУ может носить случайный характер, что в качестве примера отражено в приведенной ниже схеме.
Единственный метод лечения заключается в своевременной организации диетотерапии, которая требуется с первых дней жизни. Заключается она в резком ограничении фенилаланина, который содержится в отдельных пищевых продуктах. Таким образом, исключаются все белковые продукты. Учитывая продолжительность и полное исключение фенилаланина из пищи, возможным становится расщепление собственных белков, что ведет к истощению организма больного. По этой причине потребность в получении белка возмещается за счет аминокислотных смесей и белковых гидролизатов.
Со сменой снижения концентрации в крови фенилаланина до нормальных показателей, в рацион постепенно добавляются продукты животного происхождения. В рацион добавляются различные фрукты и сезонные овощи, растительные и животные жиры, а также углеводы. Естественно, что контроль над содержанием фенилаланина следует продолжать вести. Организм должен быть снабжен данной аминокислотой в должном объеме для обычного развития и роста малыша, однако, за исключением возможности скапливания его в тканях.
Строжайшая диета соблюдается на протяжении, как минимум, пяти лет. Что касается возраста более взрослого, то здесь в значительной степени снижается подверженность нервной системы к воздействию, оказываемому фенилаланином, а также воздействию, оказываемому продуктами его распада. При соблюдении требуемых мер к 12-14 годам ребенок сможет свободно перейти к обычному питанию.
Примечательно, что медикаментозное лечение при данном заболевании носит синдромный характер. В него включено применение препаратов, ориентированных на устранение судорог, в том числе и препаратов, которые оказывают стимулирующее воздействие на интеллектуальную деятельность. В обязательном порядке детям назначается курс лечебной физкультуры и массажа. Дополнительно назначаются уроки, способствующие развитию логики.
Диагностирование фенилкетонурии производится педиатром и генетиком на основании специальных анализов в комплексе с общей характерной симптоматикой.
Все ли корректно в статье с медицинской точки зрения?
Ответьте только в том случае, если у вас есть подтвержденные медицинские знания
Заболевания со схожими симптомами:
Отек головного мозга – опасное состояние, характеризующееся чрезмерным скоплением экссудата в тканях органа. Как следствие, постепенно увеличивается его объем и растёт внутричерепное давление. Все это приводит к нарушению обращения крови в органе и к отмиранию его клеток.
Переутомление – состояние, с которым сегодня часто сталкиваются не только взрослые, но и дети. Оно характеризуется пониженной активностью, сонливостью, нарушением внимания и раздражительностью. Причём многие люди считают, что переутомление – несерьёзная проблема, и что достаточно хорошо отоспаться, чтобы оно прошло. На самом же деле избавиться от такого нарушения невозможно длительным сном. Все наоборот — постоянное желание спать и неспособность восстановить силы после сна – это основные симптомы переутомления.
Аденома паращитовидной железы представляет собой небольшое доброкачественное образование размером от 1 до 5 см, которое может самостоятельно синтезировать паратгормон, вызывая у человека симптомы гиперкальциемии. Паращитовидные железы располагаются на задней поверхности щитовидки, и их основное назначение заключается в продуцировании паратиреоидного гормона, принимающего участие в кальциево-фосфорном обмене в организме. Аденома приводит к тому, что паратиреоидного гормона начинает вырабатываться больше, чем необходимо, что и обуславливает симптомы этого заболевания.
Гидроцефалия, которую также принято определять в качестве водянки головного мозга, представляет собой заболевание, при котором происходит увеличение объема желудочков в головном мозге, причем зачастую – до весьма внушительных размеров. Гидроцефалия, симптомы которой проявляются по причине избыточной выработки ликвора (спинномозговой жидкости между сообщающимися желудочками мозга) и его скопления в области полостей головного мозга, преимущественно возникает у новорожденных, однако имеет это заболевание и место в заболеваемости других возрастных категорий.
Гидроцефалия (син. водянка) головного мозга у детей – это болезнь, характеризующаяся тем, что во внутренних его полостях и под мозговыми оболочками собирается чрезмерное количество ликвора, что также носит название спинномозговая жидкость. Причин формирования недуга достаточно много, причём они могут отличаться в зависимости от того, в каком возрасте сформировалась патология. Наиболее часто в качестве провоцирующих факторов выступают инфекционные и онкологические процессы, врождённые пороки развития и родовые травмы.
Фенилкетонурия – это сложное генетическое заболевание, в основе которого лежит нарушение обмена аминокислот. В результате в организме не происходит трансформации жизненно важной аминокислоты фенилаланина в тирозин.
Эта болезнь достаточно распространена, а выявляется она в среднем в одном случае на 8 тысяч новорожденных. Поскольку организм при таком генетическом нарушении не получает тирозина, это обуславливает множество патологий. Ведь это вещество воздействует на работу гипофиза, щитовидки и надпочечников, а также препятствует откладыванию жира в организме и уменьшает аппетит.
Впервые фенилкетонурия у детей была выявлена в 30-е годы прошлого века ученым А. Феллингом. Первые симптомы заболевания проявляются практически сразу же после рождения, а дальнейшее его прогрессирование может привести к тяжелым осложнениям, в том числе поражению головного мозга, нарушениям развития у ребенка психики и движения, а также токсическому повреждению нервной системы.
Причины фенилкетонурии обусловлены наследственной предрасположенностью. Однако заболевание передается только в том случае, когда оба родителя являются носителями измененного гена.
Этот ген встречается только у двух процентов населения земного шара, при этом человек может быть абсолютно здоров. Однако если мужчина и женщина, являющиеся его носителями, вступают в брак, то они могут передать его своему ребенку. Даже в этом случае вероятность рождения малыша с генетическими нарушениями составляет только 25 процентов.
Также фенилкетонурия может быть обусловлена близкородственным браком, поскольку в этом случае родители могут являться носителями мутированного гена, который кодирует фермент фенилаланин-4-гидроксилаз. Фенилаланин в организме больного человека выводится очень медленно, поэтому при его постоянном поступлении он поражает нервную систему.
Если заболевание было выявлено своевременно, то качество и продолжительность жизни такого ребенка ничем не отличается от других людей. Однако при некачественной диагностике и отсутствии терапии она значительно сокращается.
Симптомы фенилкетонурии появляются у ребенка сразу же после рождения. Среди них можно выделить:
- Усталость и вялость;
- Беспокойство;
- Отсутствие интереса к окружающему миру;
- Аллергический дерматит и другие кожные заболевания;
- Судороги;
- Повышенное беспокойство и раздражительность;
- Срыгивания после еды;
- Бледность кожных покровов;
- Нарушение тонуса мышц;
- Специфический запах мочи.
В старшем возрасте симптомы фенилкетонурии проявляются в виде задержки психического и физического развития, а при проведении обследования врачи могут обнаружить микроцефалию.
К тому же больные имеют специфические внешние особенности. У них светлая фарфоровая кожа, белые волосы и голубые глаза. Если болезнь выявлена в первые дни после рождения, то при правильно назначенном лечении и диете ребенок может сохранить шансы на здоровую жизнь.
Новорожденный с этой патологией рождается абсолютно здоровым, а ухудшение состояния наступает впоследствии, поскольку организму не хватает вещества тирозина. Поэтому при раннем выявлении заболевания головной мозг не поражается, а ребенок растет аналогично своим сверстникам.
В том случае, когда фенилкетонурия после рождения не была установлена, а ребенок длительное время употреблял продукты с большим содержанием фенилаланина, наступают необратимые последствия. При его накоплении в жидкостях организма и тканях происходит токсическое отравление нервной системы и головного мозга, а в тяжелых случаях может развиться идиотия.
В возрасте после двух лет у ребенка могут проявиться следующие симптомы:
- Недержание мочи;
- Эпилептические судороги и спазмы;
- Дрожание рук;
- Уменьшение размеров черепа;
- Скованные движения, обусловленные повышенным тонусом мышц;
- Деформирование раковин ушей;
- Нарушения поведения.
При отсутствии диеты в лечении больного возможно развитие психических отклонений, которые становятся причиной инвалидности.
Заболевание может иметь разную симптоматику в зависимости от формы. Всего выделяют три типа, среди которых наиболее распространенной является первый. Он поддается лечению благодаря ограничениям в диете, а болезнь второго и третьего типа может привести к летальному исходу.

Фенилкетонурия обычно выявляется на пятый день после рождения. Для этого всем новорожденным проводят перед выпиской обследование, после кормления берут анализ крови. Ее наносят на специальные тестовые полоски, после чего осуществляется анализ, который позволяет выявить эту патологию.
Для подтверждения диагноза проводится анализ крови и мочи, в котором будет заметно повышение уровня фенилаланина. Для предотвращения симптомов заболевания ребенку назначается незамедлительное лечение и специальная диета с ограничением белковых продуктов, которая позволяет ему впоследствии вести нормальную и здоровую жизнь.
Эта генетическая болезнь также может быть диагностирована в процессе визуального осмотра и сбора анамнеза. Однако этот метод использовался еще несколько десятилетий назад, сегодня самыми проверенными являются способы лабораторной молекулярно-генетической диагностики.

Лечение фенилкетонурии проводится пожизненно, однако наиболее важным периодом является детский возраст до 18 лет. В это время наиболее активно развивается нервная система, а накопление фенилаланина в организме может привести к ее токсическому поражению.
Однако только специальная диета позволит человеку вести здоровую и полноценную жизнь вплоть до самой старости. Врачи таким пациентам советуют продолжать соблюдать диету на протяжении всей жизни, чтобы улучшить ее качество.
На первом году жизни ребенок должен питаться специальными смесями с достаточным количеством незаменимых аминокислот, витаминов и микроэлементов. К ним относятся Фенил Фри, ХР Аналог, Афенилак, MIDмил ФКУ.
Назначить определенную молочную смесь врач может только на основании результатов анализов. В дополнении к искусственным молочным смесям мать должна кормить ребенка грудью для укрепления его иммунной системы, однако дозировку молока должен подбирать специалист на основании проведенных анализов и обследований.
Для детей после одного года для питания помимо прикорма могут быть использованы Изифен, ХР Максамейд и ХР Максамум, а также П-АМ. Эти продукты не содержат фенилаланина, поэтому они не вызывают нарушений в работе внутренних органов и прекрасно перевариваются.
Диета для взрослых и детей при фенилкетонурии назначается сразу же после проведения генетического анализа. В этом случае питание должно быть богато растительными белками, витаминами и микроэлементами. К полностью разрешенным продуктам относятся – картошка, сахар, растительные масла, а также зеленый и черный чай.
Среди запрещенных продуктов можно выделить:
- Мясные и рыбные блюда, а также птица;
- Молочные продукты: сметана, йогурт, сыр, молоко и творог;
- Куриные яйца;
- Мука и макаронные изделия;
- Горох и другие бобовые;
- Кукуруза;
- Шоколад и орехи;
- Желатин.
К разрешенным продуктам, которые необходимо употреблять в ограниченном количестве, относятся – хлеб, сливочное масло, овощи и фрукты, рис, мед, а также щербеты. Также важно, чтобы в организм человека поступали продукты, богатые тирозином.
Это вещество необходимо для полноценного функционирования организма, а в больших количествах оно содержится в грибах и других растительных продуктах. Такое лечение фенилкетонурии позволит пациенту прожить долгую полноценную жизнь.
В последние годы специалистами был разработан препарат, содержащий растительную фенилаланингидроксилазу. Он позволяет контролировать содержание фенилаланина в органах и тканях, не давая ему накапливаться в больших количествах.
Фенилкетонурия является опасным и сложным генетическим заболеванием, поэтому ее лечение народными средствами не проводится.Однако возможно использование травяных сборов и настоев для улучшения общего состояния здоровья.
Они могут снять раздражительность и беспокойство у новорожденных, а также восполнить нехватку витаминов и микроэлементов, которые связаны с жесткими ограничениями при диете. Больные могут употреблять мяту, ромашку, календулу, мелиссу, боярышник, пустырник. При этом орехи и сухофрукты смогут укрепить иммунную систему. Использование всех лекарственных препаратов и трав необходимо согласовывать с врачом.

Фенилкетонурия у грудничков и новорожденных не может быть предупреждена, поскольку эта болезнь обусловлена генетическим нарушением. Однако для предупреждения опасных последствий болезни необходима ее ранняя диагностика и впоследствии тщательная диета.
При выявлении этого заболевания у одного ребенка в семье необходимо проводить перинатальную диагностику заболевания при последующей беременности, а также возможна медико-генетическая консультация.
Фенилкетонурия — это наследственное заболевание, которое характеризуется нарушением белкового обмена.
Впервые это заболевание обнаружили в 1934 году. Фенилкетонурия наследуется по так называемому аутосомно-рецессивному типу, то есть у совершенно здоровых родителей (носителей) могут родиться больные фенилкетонурией дети.
Существует 3 типа этого заболевания.
Фенилкетонурия первого типа – характеризуется дефицитом в организме фермента фенилаланин-4-гидроксилаз. Благодаря этому ферменту аминокислота фенилаланин превращается в тирозин. Чаще всего наследуется фенилкетонурия именно этого типа (в 98% случаев).
Фенилкетонурия второго типа – характеризуется недостатком такого фермента как дигидроптеридинредуктаза. Больные фенилкетонурией второго типа страдают судорогами и умственной отсталостью. Этот тип фенилкетонурии у детей встречается довольно редко (1-2%), но обычно приводит к смертельному исходу в 2-3-летнем возрасте.
Фенилкетонурия третьего типа – характеризуется дефицитом тетрагидробиоптерина. Симптомы фенилкетонурии этого типа включают в себя умственную отсталость вследствие микроцефалии – уменьшения объема мозга.
Основными причинами развития фенилкетонурии у детей считаются мутации гена, находящегося на 12 хромосоме. Родственные браки повышают риск возникновения аномалии.
При дефиците определенных ферментов наблюдается увеличение в крови производных фенилаланина, которые оказывают отравляющее действие на нервную систему ребенка. Фенилкетонурия наследуется примерно одинаково как мальчиками, так и девочками.
Симптомы фенилкетонурии проявляются не сразу. В большинстве случаев, заподозрить фенилкетонурию у детей сразу после рождения практически невозможно. На вид ребенок выглядит здоровым, рождается в срок и с нормальным весом. Но уже через несколько недель проявляются симптомы фенилкетонурии. Основным признаком заболевания считается сильная рвота. В период с двух до шести месяцев, как мама, так и лечащий доктор могут заметить отставание ребенка в психическом и физическом развитии.
Дети, больные фенилкетонурией, позже остальных сверстников начинают сидеть и ходить. Также явным симптомом фенилкетонурии является повышенная потливость с характерным «мышиным» запахом пота. Наблюдаются судороги, раздражительность, вялость, капризность и плаксивость, уменьшение размера головы, высыпания на коже. У детей, больных фенилкетонурией, поздно прорезываются зубы.
С развитием фенилкетонурии повышается мышечный тонус, что характеризуется определенной позой у ребенка, которую еще называют «поза портного» (согнутые в суставах руки и ноги).
 Единственным лечением фенилкетонурии является соблюдение диеты, которой необходимо придерживаться более десяти лет после постановки диагноза. Дети, больные фенилкетонурией, не могут усваивать большое количество фенилаланина, поэтому имеются определенные нормы его потребления, которые зависят от возраста. Ребенок до двух месяцев может потреблять не более 60 мг/кг массы фенилаланина, а дети старше шести лет – не более 10-15 мг/кг.
Единственным лечением фенилкетонурии является соблюдение диеты, которой необходимо придерживаться более десяти лет после постановки диагноза. Дети, больные фенилкетонурией, не могут усваивать большое количество фенилаланина, поэтому имеются определенные нормы его потребления, которые зависят от возраста. Ребенок до двух месяцев может потреблять не более 60 мг/кг массы фенилаланина, а дети старше шести лет – не более 10-15 мг/кг.
Что касается грудного вскармливания, то тут необходимо придерживаться строгих правил. Мама должна контролировать количество грудного молока, которое выпивает ребенок. Поэтому для кормления необходимо использовать только сцеженное молоко и в количестве, которое разрешено для определенного возраста ребенка. Для этого существуют специальные таблицы с нормами потребления фенилаланина, а также формулы расчета количества грудного молока в день. Докармливают ребенка специальными смесями, которые не содержат фенилаланина.
Введение прикорма у детей с фенилкетонурией начинают с фруктовых и ягодных соков. В качестве твердой пищи предлагают ребенку овощные пюре без добавления молока. Используют безбелковые крупы и безмолочные каши на основе рисовой или кукурузной муки.
Лечение фенилкетонурии посредством питания включает в себя отказ или очень ограниченное потребление таких продуктов как рыба, мясо, хлебобулочные изделия, колбасы, творог, яйца, шоколад, бобовые, орехи и крупы. Эти продукты содержат большое количество белка. Фрукты и овощи вводят в рацион с учетом подсчета количества в них фенилаланина.
Лекарственное лечение фенилкетонурии предполагает прием витаминов, препаратов кальция, железа и фосфора. Также показан прием препаратов для улучшения мозгового кровообращения, микроциркуляции и тканевого обмена. Рекомендуют занятия лечебной физкультурой и массаж.
источник
Природа не только мудра, но и щедра. Она одаривает людей многими радостями, исполняя их заветные желания иметь здоровых детей, хотя время от времени преподносит «сюрпризы», застающие молодые семьи врасплох. Один из них принадлежит к сфере генетических закономерностей. Без наследственности и изменчивости, благодаря которым у потомства появляются новые признаки, невозможна была бы эволюция жизни на Земле, однако при этом человечество отягощается грузом разнообразных мутаций, то есть изменений в наследственных структурах. Болезни, передающиеся потомству по наследству, являются частью общей наследственной изменчивости человека. Они выявляются, у новорожденных во внешне вполне благополучных семьях. В этих случаях практически здоровые родители имеют патологический задаток того или иного наследственного заболевания, о чем они не знают.
Редкое заболевание фенилкетонурия (ФКУ) — одна из форм наследственных дефектов обмена аминокислот. В нашей стране частота этого заболевания невелика: один больной ребенок приходится на семь тысяч здоровых новорожденных. Малыш рождается с генетическим дефектом, из-за которого аминокислота фенилаланин, поступающая в организм с пищевым белком, не может превращаться в тирозин, как это бывает в норме. В результате фенилаланин и его производные накапливаются в тканях и органах малыша, оказывая токсическое воздействие на нервную систему. Если не диагностировать ФКУ в период новорожденности и, соответственно, ничего не предпринимать, болезнь приведет к весьма серьезным последствиям: у ребенка разовьется выраженная умственная отсталость — олигофрения, вплоть до крайней степени — идиотии. При отсутствии своевременного лечения больные на всю жизнь останутся глубокими инвалидами, так как повернуть болезнь «вспять» невозможно. Однако недаром говорится, что береженого Бог бережет. Поэтому родители каждого новорожденного должны понимать, как важно вовремя обследовать ребенка, чтобы не упустить время начала лечения в случае установления диагноза ФКУ.
Больной ребенок с ФКУ может родиться только в той семье, где родители практически здоровы, но оба являются носителями патологического задатка, в данном случае — носителями гена ФКУ.
Определить носительство гена ФКУ у родительской пары возможно при генетическом обследовании на гетерозиготное носительство, проводимое в некоторых федеральных медико-генетических центрах страны. Если в семье уже есть больной ребенок с ФКУ, носительство гена ФКУ у родителей очевидно.
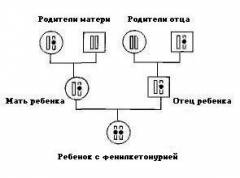
Однако даже если оба родителя являются носителями гена ФКУ, их дети не обязательно будут больны. Если принять за 100% всех детей, которые гипотетически могут родиться в данной семье, можно говорить о следующем риске возникновения заболевания ФКУ:
- риск рождения больных детей с ФКУ составляет 25%;
- риск рождения детей, являющихся, подобно их родителям, носителями гена ФКУ составляет 50%;
- в остальных 25% случаев родятся здоровые дети (см. рис.).
Известно, что носительство мутантного гена ФКУ среди населения составляет 2 —3%. Однако, как было сказано выше, риск рождения ребенка с ФКУ появится только в том случае, если оба родителя являются носителями патологического гена. Поэтому данное заболевание встречается довольно редко.
Ребенок с ФКУ рождается без каких-либо проявлений заболевания. Однако с началом кормления, при поступлении в организм белка грудного молока или его заменителей возникают первые микросимптомы, трудно распознаваемые не только родителями, но и педиатрами.
Так, в периоде новорожденности до начала лечения у ребенка с ФКУ возможны необоснованная вялость или беспокойство; обращают на себя внимание рассеянный, блуждающий взгляд, отсутствие улыбки, слабое двигательное оживление. К 6 месяцам у него выявляется задержка психомоторного развития: он перестает активно реагировать на происходящее; утрачивает способность узнавать мать; не переворачивается на живот; не пытается сесть.
Во втором полугодии жизни родители уже не могут не заметить непонимание речи взрослого, неумение выражать голосом и мимикой ребенка свои переживания. У детей старше трех лет нарастают умственная отсталость, возбудимость, повышенная утомляемость; нарушается поведение, что проявляется в расторможенности, психотических расстройствах.
Часто у нелеченых больных ФКУ моча имеет своеобразный «мышиный» запах. Иногда возникают судорожные приступы различной степени выраженности; экзематозные изменения на коже.
С первых дней после появления на свет здоровый ребенок должен очень быстро набирать темп развития, приобретая разнообразные физиологические навыки. Следите за его психомоторным развитием, постоянно консультируйтесь у врача-педиатра.
Из многочисленных наследственных заболеваний обмена веществ (а их насчитывается не менее 700) фенилкетонурия — самое «благоприятное», поскольку при ранней диагностике возможна полная реабилитация больного и его полноценная адаптация к социальной жизни, чего нельзя достигнуть при многих других видах наследственной патологии.
К сегодняшнему дню вопрос ранней диагностики у новорожденных заболевания ФКУ решен. В течение последних 10 лет отшлифовывалась методика массового обследования (скрининга) всех новорожденных в нашей стране по выявлению заболевания фенилкетонурией. Приказом Минздрава РФ №316 от 30.12.1993 г. организовано обеспечение массового скрининга новорожденных на фенилкетонурию на всей территории России. На службе у программы скрининга находятся все родовспомогательные учреждения страны, детские районные поликлиники, более 80 медико-генетических центров, консультаций и кабинетов.
Для раннего выявления ФКУ всем новорожденным проводится скрининг-тест (анализ крови ). Это самый надежный способ позаботиться о здоровье малыша с первых дней его рождения.
Скрининг-тест заключается в следующем:
- У новорожденного не позднее чем на четвертый-пятый день его жизни натощак (через 3 часа после кормления) берется несколько капель крови из пяточки.
- Кровь наносится на специальный бумажный бланк, выданный лабораторией скрининга в медико-генетическом центре региона по месту рождения ребенка. Необходимо, чтобы капли крови были нанесены на бланк тремя насквозь пропитанными кровью кружками диаметром не менее 12 мм.
- Бланк с кровью как можно быстрее должен быть возвращен в лабораторию скрининга для проведения анализа крови на содержание в ней аминокислоты — фенилаланина (ФА).
- Анализ крови на ФА выполняется через сутки после поступления бланка в лабораторию.
- Результат скрининг-теста заносится в обменную карту ребенка в роддоме в виде штампа: «На ФКУ и ВГ 1 обследован».
При выписке из роддома обязательно проверьте, был ли проведен скрининг-тест на ФКУ.
Если роды происходили вне родильного дома (в обычной больнице, дома и т.д.) и скрининг-тест не производился, родителям самостоятельно без промедления следует обратиться в региональную медико-генетическую консультацию.
В Москве скрининг-тест и консультация врача-генетика проводятся в Московском центре неонатального скрининга по адресу: 119334, 5-й Донской проезд, д.21 а. Телефоны: 952-22-28, 954-41-27.
Если скрининг-тест окажется положительным, вас оповестят и пригласят на консультацию к врачу-генетику. При повторном положительном результате анализа на ФА следует немедленно начать лечение ребенка.
Диагноз ФКУ должен быть поставлен (или отвергнут) не позднее трехнедельного возраста ребенка!
Родителям не следует поддаваться панике: вероятность того, что заболевание будет обнаружено именно у вашего ребенка, очень невелика, но даже если это случится, — не волнуйтесь: развитие болезни можно предотвратить при своевременно начатом лечении.
Единственным эффективным методом лечения больных ФКУ является специализированная диетотерапия с момента установления диагноза. Смысл диетического лечения сводится к резкому ограничению поступающего с пищей белка животного происхождения и замене его специализированными лечебными продуктами. Лечебный продукт — это сухая смесь аминокислот без фенилаланина, являющаяся практически единственным источником пищевого белка в рационе, необходимым для роста и развития ребенка. Родители детей, больных ФКУ, получают лечебные продукты в медико-генетической консультации бесплатно.
В случае выявления заболевания фенилкетонурии у новорожденного родители немедленно получают квалифицированную консультативную помощь и специализированную литературу у врача-генетика в медико-генетическом центре, консультации или кабинете по месту жительства. Госпитализации ребенка не требуется.
Течение беременности у женщины, являющейся носительницей гена ФКУ ничем не отличается от течения беременности здоровой женщины.
Желаю вам благополучных родов и исполнения заветной мечты — рождения здорового ребенка.
источник
Фенилкетонурия (ФКУ). Причины, симптомы, диагностика и лечение фенилкетонурии. Лечение фенилкетонурии: диета и питание.
Фенилкетонурия (ФКУ) – довольно редкое наследственное заболевание, связанное с нарушением обмена аминокислот. Организм больного фенилкетонурией человека не способен расщеплять аминокислоту фенилаланин, которая поступает с белковой пищей. В результате этого, в тканях накапливаются соединения, отравляющие нервную систему и головной мозг в частности. Развивается умственная отсталость (малоумие), вплоть до идиотии. В связи с этим болезнь получила и другое название – фенилпировиноградная олигофрения.
Однако из всех наследственных заболеваний фенилкетонурия, единственное, которое удается полностью нейтрализовать. Сегодня ребенка, рожденного с признаками ФКУ, можно вырастить абсолютно здоровым. Обезопасить мозг малыша удается с помощью специальной диеты, о которой мы расскажем ниже.
В разных странах частота этого заболевания отличается в разы. В России рождается один больной ребенок на 10 000. В некоторых регионах Великобритании этот показатель в два раза выше – 1:5000. Дети на Африканском континенте практически не болеют фенилкетонурией. Среди больных количество девочек почти в два раза превышает количество мальчиков.
 Заболевание наследуется только в том случае, если оба родителя передали ребенку склонность к болезни, и поэтому встречается довольно редко. У двух процентов людей есть измененный ген, который отвечает за развитие болезни. При этом человек остается полностью здоровым. Но когда мужчина и женщина, носители мутировавшего гена, вступают в брак и решают завести детей, то вероятность того, что малыши будут страдать от фенилкетонурии, составляет 25%. А возможность того, что дети будут носителями патологического гена ФКУ, но сами останутся практически здоровыми, составляет 50%.
Заболевание наследуется только в том случае, если оба родителя передали ребенку склонность к болезни, и поэтому встречается довольно редко. У двух процентов людей есть измененный ген, который отвечает за развитие болезни. При этом человек остается полностью здоровым. Но когда мужчина и женщина, носители мутировавшего гена, вступают в брак и решают завести детей, то вероятность того, что малыши будут страдать от фенилкетонурии, составляет 25%. А возможность того, что дети будут носителями патологического гена ФКУ, но сами останутся практически здоровыми, составляет 50%.
Причина возникновения этого заболевания связана с тем, что в печени человека не вырабатывается особый фермент – фенилаланин-4-гидроксилаза. Он отвечает за превращение фенилаланина в тирозин. Последний входит в состав пигмента меланина, ферментов, гормонов и необходим для нормальной работы организма.
При ФКУ фенилаланин, в результате побочных путей обмена, превращается в вещества, которых не должно быть в организме: фенилпировиноградную и фенилмолочную кислоты, фенилэтиламин и ортофенилацетат. Эти соединения накапливаются в крови и оказывают комплексное действие:
- нарушают процессы жирового обмена в мозге
- вызывают дефицит нейромедиаторов, которые передают нервный импульс между клетками нервной системы
- оказывают токсическое действие, отравляя мозг
Это вызывает значительное и необратимое снижение интеллекта. У ребенка быстро развивается умственная отсталость – олигофрения.
 Дети с ФКУ рождаются абсолютно здоровыми. Поэтому, если в течение первых дней жизни выявить заболевание и придерживаться диеты, то удается предотвратить разрушение мозга ребенка. При этом, никакие признаки заболевания не появляются. Малыш развивается и растет, как и его сверстники.
Дети с ФКУ рождаются абсолютно здоровыми. Поэтому, если в течение первых дней жизни выявить заболевание и придерживаться диеты, то удается предотвратить разрушение мозга ребенка. При этом, никакие признаки заболевания не появляются. Малыш развивается и растет, как и его сверстники.
Если же момент упущен, и ребенок употребляет в пищу белковые продукты, богатые фенилаланином, то начинают проявляться симптомы поражения центральной нервной системы. Поначалу изменения у больных фенилкетонурией незначительны. Их трудно заметить даже опытному педиатру. Это слабость и беспокойство. Малыш не улыбается и мало двигается.
К шести месяцам задержка развития становится более заметной. Ребенок слабо реагирует на происходящее, не узнает мать, не пытается сесть и перевернуться. Фенилаланин и его производные выводятся из организма с мочой и потом. Они вызывают специфический «мышиный» или затхлый запах.
Годовалый ребенок не умеет выражать голосом свои эмоции и переживания, имеет невыразительную мимику, не понимает речь родителей.
В возрасте трех лет и старше симптомы фенилкетонурии нарастают. У детей наблюдается повышенная возбудимость, утомляемость, нарушения поведения, психотические расстройства, умственная отсталость. Если не заниматься лечением фенилкетонурии, то состояние больного будет ухудшаться.
 В том случае, если есть подозрение, что один или оба родителя являются носителями гена ФКУ, то определить это можно в федеральных медико-генетических центрах. Для установления этого факта проводится генетическая экспертиза.
В том случае, если есть подозрение, что один или оба родителя являются носителями гена ФКУ, то определить это можно в федеральных медико-генетических центрах. Для установления этого факта проводится генетическая экспертиза.
На сегодняшний день все новорожденные дети массово обследуются на наличие фенилкетонурии. На территории России этот вопрос регламентирует приказ Минздрава РФ №316 от 30.12.1993 г. Процедура получила название неонатальный скрининг и является эффективным способом выявления наиболее распространенных наследственных заболеваний, среди них и ФКУ.
Массовое обследование новорождённых это простой и достоверный метод диагностики. В роддоме у каждого ребенка берут несколько капель периферической крови из пяточки. Это делается натощак, через три часа после кормления. У доношенных детей анализ берут на четвертый день жизни, а у недоношенных на седьмой. У тех новорожденных, которые появились на свет не в родильных домах, важно взять анализ на протяжении первых трех недель.
Кровь наносят на специальный тест-бланк, который потом отправляют в лабораторию для проведения генетического исследования. Там на протяжении суток проводится анализ крови на содержание в ней аминокислоты — фенилаланина. Результаты теста заносятся в обменную карту ребенка в виде штампа: «На ФКУ и ВГ обследован».
В том случае, если в анализе обнаруживают измененный ген, то родителей с ребенком приглашают в медико-генетический центр для обследования. Для того, чтобы подтвердить или опровергнуть диагноз назначаются дополнительные исследования:
- в сухом пятне крови
- в сыворотке крови
- потовый тест
- копрограмма
- ДНК-диагностика
В любом случае родители должны понимать, что при своевременно начатом лечении и соблюдении диеты удается полностью предотвратить развитие болезни.
На сегодняшний день в нашей стране единственным эффективным методом лечения является диетотерапия. Разрабатываются препараты, которые позволят контролировать уровень фенилаланина в крови без соблюдения диеты. В этом направлении есть значительные успехи, но в продаже такие лекарственные средства появятся не раньше чем через 5-7 лет.
Постоянно идет работа над поиском новых средств и методов борьбы с болезнью.
- Перспективным направлением считается использование растительного фермента фенилаланинлиазы, который будет расщеплять излишки фенилаланина в организме.
- Ученые возлагают большие надежды на генотерапию с использованием вирусного фактора, которая позволит вылечить больной ген и полностью избавиться от проблемы.
- Практикуется введение гена фенилаланингидроксилазы прямо в пораженные клетки печени.
Но в нашей стране эти разработки пока не используются. Лечение с помощью диеты – основная помощь больным фенилкетонурией. Ограничить употребление белка необходимо с самого рождения и до половой зрелости. За ростом и развитием ребенка постоянно наблюдают врачи: педиатр и невролог. Специалисты корректируют количество белков, чтобы они соответствовали возрасту и нагрузкам ребенка.
Некоторые формы фенилкетонурии поддаются лечению тетрагидробиоптерином, который является составляющей частью недостающего фермента фенилаланин-4-гидроксилазы. Атипичные формы ФКУ не лечатся с помощью диеты и требуют регулярного приема тетерагидробиоптерина или его заменителей.
 Для того, чтобы нервные клетки ребенка не подвергались токсическому воздействию фенилаланина и его производных, нужно полностью исключить из рациона животные белки. Если это сделать на первых неделях жизни, то мозг останется полностью здоровым. Если же начинать ограничивать белок в более позднем возрасте, то задержку развития удается несколько приостановить. Но вернуть здоровье нервной системе и устранить изменения в нервных клетках уже не удастся.
Для того, чтобы нервные клетки ребенка не подвергались токсическому воздействию фенилаланина и его производных, нужно полностью исключить из рациона животные белки. Если это сделать на первых неделях жизни, то мозг останется полностью здоровым. Если же начинать ограничивать белок в более позднем возрасте, то задержку развития удается несколько приостановить. Но вернуть здоровье нервной системе и устранить изменения в нервных клетках уже не удастся.
Соблюдать диету требуется до 16-18 лет. Это обязательное условие. Желательно контролировать количество животных белков и в дальнейшем.
Если женщина, у которой в детстве были обнаружены признаки ФКУ, планирует забеременеть, то ей обязательно нужно вернуться к диете без фенилаланина. Таких ограничений необходимо придерживаться до зачатия, во время беременности и кормления грудью.
Все необходимые для роста и развития аминокислоты поступают в организм из специализированных лечебных продуктов. Обычно они представляют собой порошок – сухую смесь аминокислот. Их родителям больного малыша выдают бесплатно в медико-генетической консультации.
Грудные дети получают специальные смеси полностью очищенные от лактозы на основе гидролизата молочного белка.
Питательные средства, которые должны заменить детям натуральные белковые продукты, содержат:
- пептиды (расщепленные ферментами молочные белки);
- свободные аминокислоты (тирозин, триптофан, цистин, гистидин и таурин).
В России применяются такие смеси: Афенилак, Аналог-СП, Мдмил-ФКУ-0. Они представляют собой порошки, которые необходимо разводить кипяченой водой или сцеженным грудным молоком, согласно инструкции. В результате получается жидкая смесь или «сметанка». Такой прикорм вводят постепенно, в течение 2-5 дней под наблюдением врача.
Специальными диетическими продуктами для пополнения запасов белка у детей разного возраста также являются: “Берлафен“, “Циморган“, “Минафен“, “Апонти“.
Детей, больных ФКУ, можно кормить грудью. Но при этом кормящей матери нужно придерживаться специальной диеты.
В диете детей дошкольного и школьного возраста полностью исключают из меню белковые продукты. В списке разрешенных продуктов – овощи, фрукты, изделия из крахмала, растительные масла. При составлении дневного меню необходимо строго придерживаться возрастных норм фенилаланина.
| Возраст ребенка | Суточное количество фенилаланина (мк/кг массы тела) |
| Младше 2 мес. | 60 |
| 2-3 мес. | 60-55 |
| 3-6 мес. | 55-45 |
| 6-12 мес. | 45-35 |
| 1-1,5 года | 35-30 |
| 1,5-3 года | 30-25 |
| 3-6 лет | 25-15 |
| Старше 6 лет | 15-10 |
Необходимо помнить, что для растущего организма полноценное питание жизненно необходимо. Так в сутки ребенку требуется 120 мг тирозина на каждый килограмм массы. Поэтому дети и подростки с таким диагнозом должны получать аминокислоты для построения клеток и роста из дополнительных источников. Также обязательно назначают витаминно-минеральный комплекс. Особенно важно, чтобы ребенок получал норму витаминов С, В6 и B1, фолиевой кислоты, железа, кальция и магния. Количество калорий должно быть увеличено на 30% по сравнению с дневной нормой сверстников.
Разделяют три группы натуральных продуктов. В основе классификации лежит количество в них фенилаланина:
- Красный список – продукты, которые необходимо полностью исключить из рациона.
- Оранжевый список – разрешены в небольших количествах под строгим контролем.
- Зеленый список – могут употребляться без ограничений.
| Красный список | Оранжевый список | Зеленый список |
| Все виды мяса | Молочные продукты | Фрукты |
| Колбасные изделия | Рис и кукуруза | Ягоды |
| Все виды рыбы | Овощи (картофель, капуста) | Зелень |
| Морепродукты | Овощные консервы | Овощи |
| Яйца | Рисовая, кукурузная мука | |
| Сыры | Крахмал и саго | |
| Творог | Сахар и варенье | |
| Орехи | Мед | |
| Хлеб и хлебобулочные изделия | Сливочное и растительное масло, топленый жир | |
| Кондитерские изделия | ||
| Крупы и хлопья | ||
| Продукты из сои | ||
| Поп-корн | ||
| Аспартам |
Промышленность выпускает еще две группы продуктов:
- искусственные низкобелковые продукты, специально для диетического питания (хлеб, печенье, макароны)
- готовые пюре для детского питания на основе фруктов.
На основе этих продуктов можно составить полноценное меню, готовить ребенку вкусные, полезные и разнообразные блюда.
Родителям ребенка, больного ФКУ, важно уметь составлять диету и правильно рассчитывать количество фенилаланина. Для этого необходимо иметь под рукой весы, которые дают возможность взвешивать до десятой доли грамма.
 Необходимо контролировать количество фенилаланина. Оно должно находиться в границах 3–4 мг% или 180–240 мкмоль/л.
Необходимо контролировать количество фенилаланина. Оно должно находиться в границах 3–4 мг% или 180–240 мкмоль/л.
Для определения необходимо сделать анализ крови в лаборатории. До трехмесячного возраста это делают еженедельно.
Постепенно врач снижает количество анализов. С тех месяцев до года – один раз в месяц, с года до трех лет – один раз в два месяца. После трех лет частота проверок снижается до одного раза в три месяца. Существует специальная схема, но специалист может изменить ее, исходя из состояния больного.
Делать анализ желательно утром натощак. От качества и регулярности такого контроля и своевременных исправлений в диете зависит сохранение интеллекта.
Новорожденные с диагнозом ФКУ ничем не отличаются от здоровых детей. И если болезнь вовремя выявить и остановить ее развитие, то и в дальнейшем такой ребенок останется абсолютно здоровым.
Как выглядят больные фенилкетонурией? 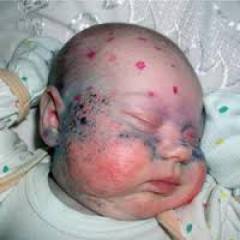
При рождении младенцы, больные фенилкетонурией, ничем не отличаются от остальных детей. Но на втором месяце жизни начинают проявляться изменения:
- посветление волос и радужки глаза из-за недостатка пигмента меланина
- чрезмерная прибавка в весе
- быстро зарастает большой родничок
- суховатая кожа
- шелушение, сыпь и экзема
- частая рвота
- моча и пот с характерным «мышиным» запахом
На втором полугодье дети, не получающие лечение, перестают узнавать мать, не могут фиксировать взгляд на одном предмете, не реагируют на яркие игрушки, не садятся и не переворачиваются, становятся раздражительными. В возрасте 2-3 года отмечаются такие особенности:
- появляются судороги и спазмы
- скованность движений и зажатая «поза портного», что связанно с повышенным напряжением в мышцах
- неадекватное поведение, выкрики, смех
- уменьшение размеров черепа
- деформация ушных раковин
- дрожание пальцев рук
- недержание мочи
- выступающая вперед нижняя челюсть
Внешние признаки болезни выражены незначительно, но при отсутствии диеты развиваются сильные психические отклонения, приводящие к инвалидности.
Для того чтобы обеспечить детей всеми необходимыми веществами в достаточном количестве разработаны специальные смеси на основе незаменимых и заменимых аминокислот. В их состав также входят витамины и все необходимые микроэлементы.
Для детей до одного года рекомендуют:
- Афенилак 13, Афенилак 15 от компании «Нутритек», Россия;
- MIDмил ФКУ 0 (Hero, Испания);
- ХР Аналог («Нутриция», Голландия);
- Фенил Фри 1 («Мид Джонсон» США).
Для детей старше одного года и для взрослых:
- П-АМ 1, П-АМ 2, П-АМ 3;
- Изифен (готовый продукт), а также ХР Максамейд и ХР Максамум с нейтральным и фруктовым вкусами («Нутриция», Голландия).
Эти продукты имеют прекрасные вкусовые качества и хорошо переносятся. Они необходимы детям и взрослым с диагнозом ФКУ в периоды умственных и физических нагрузок. Смеси удобны в применении, питательны и полностью покрывают потребности организма в аминокислотах.
 На сегодняшний день в России для лечения ФКУ используют специальную безфенилаланиновую диету. Для пополнения запасов тирозина и других аминокислот, все больные дети бесплатно получают специальные препараты. Соблюдать диету желательно до 18 лет, хотя ряд врачей утверждает, что лучше делать это на протяжении всей жизни.
На сегодняшний день в России для лечения ФКУ используют специальную безфенилаланиновую диету. Для пополнения запасов тирозина и других аминокислот, все больные дети бесплатно получают специальные препараты. Соблюдать диету желательно до 18 лет, хотя ряд врачей утверждает, что лучше делать это на протяжении всей жизни.
Такой метод диетотерапии является самым дешевым и действенным. На протяжении многих лет он помогает детям с этой болезнью вырасти здоровыми. В своем развитии они ничем не уступают сверстникам. Те, кому в детстве ставили диагноз «фенилкетонурия», учатся в школе, получают высшее образование, заводят семью и рожают здоровых детей.
Подводя итоги, отметим, что фенилкетонурия это тяжелое генетическое заболевание, которое может привести к психической инвалидности. Изменения происходят в нервной системе очень быстро и имеют необратимый характер. Однако можно не допустить развития болезни. Для этого необходима ранняя диагностика и специальная диета.
- ФенилкетонурияI. Классическая и наиболее распространенная форма заболевания, описанная выше в статье. Связана с мутацией гена в 12-й хромосоме, при этом нарушается образование фермента фенилаланин-4-гидроксилазы, который превращает фенилаланин в тирозин.
- ФенилкетонурияII. При этой форме заболевания нарушение происходит в 4-й хромосоме. Нарушается выработка фермента дигидроптеридинредуктазы, который также способствует превращению фенилаланина в тирозин. Заболевание наследуется так же, как и I форма: для того, чтобы родился больной ребенок, необходимо, чтобы носителями гена были оба родителя. Распространенность фенилкетонурии II – 1 случай на 100 000 новорожденных.
- ФенилкетонурияIII. В результате генетических нарушений возникает недостаток фермента 6-пирувоилтетрагидроптеринсинтазы. Наследуется, как и две предыдущие формы заболевания. Распространенность – 1 случай на 300 000 новорожденных.
Критерии установления инвалидности при фенилкетонурии:
- При фенилкетонурии I инвалидность устанавливают только при необратимых нарушениях со стороны центральной нервной системы, которые приводят к неврологическим расстройствам и умственной отсталости.
- При фенилкетонурии II и III типа группу инвалидности устанавливают во всех случаях.
 Специальной профилактики фенилкетонурии не существует. Но некоторые мероприятия помогают правильно оценить риски, вовремя принять необходимые меры:
Специальной профилактики фенилкетонурии не существует. Но некоторые мероприятия помогают правильно оценить риски, вовремя принять необходимые меры:
- Генетическое консультирование. Необходимо людям, планирующим завести ребенка, которые больны или являются носителями неправильного гена, у которых болен хотя бы один близкий родственник или уже родился больной ребенок. Консультирование проводит врач-генетик. Он помогает разобраться, как ген, ответственный за фенилкетонурию, передавался в предыдущих поколениях, каковы риски будущего ребенка. Также генетик помогает с планированием семьи.
- Скрининг новорожденных. Анализ не помогает предотвратить заболевание, но позволяет выявить его максимально рано, пока оно еще не привело к необратимым изменениям в головном мозге.
- Консультации и диета для женщин, страдающих фенилкетонурией. Если вы женщина и страдаете ФКУ, вам следует проконсультироваться с врачом и спросить, когда лучше планировать беременность в вашем случае. Во время беременности нужно соблюдать правильную диету – это помогает предотвратить дефекты развития у ребенка.
Прогноз зависит от формы заболевания и начала лечения, соблюдения диетических рекомендаций, медико-педагогической коррекции.
При фенилкетонурии I, при своевременном начале необходимых мероприятий, прогноз, как правило, благоприятный. Ребенок растет и развивается нормально. Если опоздать с лечением и диетой, то результат будет не таким хорошим.
При фенилкетонурии II и III прогноз серьезнее. Соблюдение диеты не приносит эффекта.
источник