Талассемия (греч.слова «thalassa» — море) – генетическая болезнь рецессивного типа (не сцепленная с полом). Для талассемии характерно снижение синтеза или дисбаланс полипептидных цепей (2 a- и — цепей), из которых построен нормальный гемоглобин. По данным статистике ВОЗ заболевание распространено в тропических странах, Средиземноморье и других теплых прибрежных странах. За последние пять лет вспышки талассемии у детей начались на территориях, не свойственных ей. В Москве зарегистрировано 162 случая талассемии у детей, из них 102 – у мальчиков и 50 – у девочек. Высокий показатель заболевания в Российской федерации отмечается в Поволжье.
Талассемии – это генетические болезни кровеносной системы организма, развивающиеся при дисбалансе синтеза a — или ? — цепей белка гемоглобина.
Заболевание является приспособительной реакцией на защиту от инфицирования паразитическими микроорганизмами, передаваемыми при укусах комаров (малярия). Гетерозиготные люди, имеющие мутации в генах альфа — и бета — цепей белка, проявляют резистентность к малярийному плазмодию, возбудителю малярии. Гомозиготное носительство приводит к тяжелым нарушениям в системах органов человека (85% детей не доживают до 14 лет) или летальному исходу при рождении или внутриутробно.
При гемоглобинопатиях накапливается избыточное количество железа в организме, обусловленное уменьшенным усвоением тканями костного мозга и увеличенным всасыванием в кишечнике. Продолжаем разбираться в вопросе талассемия, что это за болезнь и какие у нее симптомы. Железо не откладывается в запас, а при помощи ферментов поступает в органы кроветворения. При избытке железа начинается гемосидероз внутренних органов — сердце, печень, кожные покровы, железы внутренней секреции (поджелудочная железа). При всех видах талассемии эритроциты изменяют свою форму.
При дальнейшем развитии талассемии у детей страдает ЦНС – отставание в развитие, нарушения речи, гипервозбудимость и гиперактивность. В пубертатном периоде происходят изменения в половом созревании (дисбаланс в гормональном фоне).
Амалу 15 лет, диагноз талассемия, фото:
Клиническая картина талассемии зависит от вида и от степени носительства заболевания – гомозиготность или гетерозиготность.
Основные клинические проявления талассемии:
- нарушения в функционировании печени, желчного пузыря и желчевыводящих протоков — желтуха, холелитиаз;
- увеличение размеров селезенки;
- нарушения различного генеза сердечно-сосудистой системы;
- нарушения в работе ЦНС – гиперактивность, гипервозбудимость, отставание в развитии у детей.
По клиническим проявлениям врачи разделяют симптомы болезни на три типа: большая, малая, минимальная. Между этими типами есть и переходные.
Большой тип, гомозиготное носительство болезни (болезнь Кули), проявляется следующими клиническими признаками:
- анемии гемолитического типа;
- гепатоспленомегалия;
- остеопороз плоских костей черепа.
Малый тип, гетерозиготное носительство проявляется клиническими признаками:
- анемии различного генеза;
- спленомегалия, гепатоспленомегалия (размеры печени увеличены меньше, чем при больших типах);
- изменение в строении плоских костей черепа.
Минимальный тип талассемии протекает бессимптомно. Клинически проявляется при паразитарных инфекциях, беременности, травмах с большой кровопотерей.
Данное заболевание не поддается лечению. При выраженных клинических проявлениях назначают лечения, уменьшающие их. При гомозиготном носительстве талассемии у детей с рождения врачи выписывают курс переливания размороженных или отмытых эритроцитов — гемотрансфузионная терапия заболевания.
Дополнительно назначают железосвязывающие препараты для профилактики гемосидероза внутренних органов у детей – десферал. Лечебную дозировку выписывает врач с учетом возраста и массы ребенка. В тяжелых случаях детям требуется хирургическое, оперативное лечение.
При гомозиготном носительстве исход заболевания неблагоприятный. 85% детей не доживают до 14 лет. При гетерозиготном носительстве исход талассемии у детей зависит от вида и количества мутаций в генах. В 45% случаях больные живут полноценной жизнью.
источник
Наследственные заболевания встречаются довольно часто в нашей жизни: одни из них не причиняют сильного вреда для организма, другие же могут привести к летальному исходу. Одним из заболеваний, передающихся по наследство, является талассемия. Попытаемся разобраться, кому она может передаться, как отразиться на человеке и можно ли с этим жить.
Талассемия – заболевание, в основе которого лежит нарушение синтеза цепей гемоглобина. Она относится к количественным гемоглобинопатиям.
Причиной талассемии являются точечные мутации или делеции в генах, кодирующих цепи гемоглобина. В результате это может привести к уменьшению синтеза или полному отсутствию одной из цепей в организме. Другая цепь образует неадекватные тетрамеры гемоглобина, что приводит к разрушению эритроцитов и гемолитической анемии.
Гемоглобин – белок содержащийся в эритроцитах, отвечающий за перенос кислорода к тканям и углекислого газа от них.
Гемоглобин (речь идёт о HbA) состоит из четырёх цепей: двух альфа-субъединиц и двух бета-субъединиц. Такой гемоглобин составляет 97% от общего содержания его в эритроцитах.
Каждая из цепей гемоглобина связывается с его небелковой частью – гемом.
Так вот при талассемии нарушается синтез одной из цепей гемоглобина: либо альфа, либо бета. По этому принципу имеется классификация талассемии на:
По степени тяжести выделяют талассемию:
- лёгкой степени;
- средней степени;
- тяжёлой степени.
Это болезнь, относящаяся к группе наследственных заболеваний крови, которые обычно выявляются в детском возрасте. Она вызвана нарушением синтеза гемоглобина, жизненно необходимого для переноса кислорода в крови. В этой статье мы расскажем о том, что за болезнь талассемия — признаки, формы, лечение, диагностика, причины заболевания.
Гемоглобин — это сложная молекула, входящая в состав эритроцитов и необходимая для доставки кислорода в ткани организма. При нарушении синтеза гема развивается анемия, и ткани организма не получают достаточного количества кислорода для нужд метаболизма.
Нормальный гемоглобин состоит из четырех белковых цепей, называемых глобинами, каждая из которых связана с несущей кислород молекулой гема. Известно два типа глобина — альфа и бета. Они соединяются в нары, образуя молекулы гема. Количество цепей примерно одинаково, поэтому они находятся в равновесии друг с другом.
При талассемии у детей нарушено образование либо альфа-цепей либо бета-цепей. Это приводит к синтезу дефектного гемоглобина. Именно недостаток нормального гемоглобина в кровотоке и приводит к развитию анемии и симптомам заболевания талассемией.
Увеличенная селезенка часто является одним из симптомов болезни. Она захватывает эритроциты, что приводит к уменьшению их количества в кровеносных сосудах и, следовательно, к усилению анемии.
Иногда удаление селезенки облегчает состояние больных детей. Но пациенты, перенесшие спленэктомию. подвержены инфекции пневмококковыми бактериями. Им необходимо сделать прививку и давать пенициллин в течение всей жизни для предотвращения инфекций.
Существует две основных формы талассемии:
- бета, при которой нарушено образование цепей В-глобина,
- альфа, при которой нарушено образование цепей аглобина.
Оба вида подразделяются на различные подтипы разной степени тяжести. Во всех своих формах, признаки альфы обычно протекают не так тяжело.
Болезнь неизлечима, поэтому больные дети могут страдать от ее последствии всю свою жизнь. Однако использование ряда методов лечения помогает пациентам справляться с ней.
Многократные переливания крови — основа лечения. После постановки диагноза детям регулярно делают переливания крови, обычно каждые 4 — 6 недель. Цель этих процедур — повысить количество разных типов клеток в крови (число форменных элементов крови) и тем самым уменьшить степень анемии. Переливания крови абсолютно необходимы для пациентов с большой бета-талассемией. Без них больные умрут.
Лечение талассемии с помощью переливания крови, позволяет корректировать степень анемии и предотвращают развитие у малышей характерных изменений костей.
Главная проблема, возникающая при многократных переливаниях крови, — образование избытка железа в организме. Это может привести к повреждению печени, сердца и других органов.
- Для снижения токсичности железа применяют вливания препарата дефероксамина.
- Детям назначают пероральный прием витамина С способствующего дальнейшему выведению железа из организма.
Лечение увеличивает среднюю продолжительность жизни больных с большой бета талассемией, но не приносит выздоровления. В некоторых случаях дети доживают до совершеннолетия. Прогноз очень неблагоприятный. Несмотря на лечение, дети с этим типом талассемии, крайне редко доживают до возраста половой зрелости.
Известны три формы: малая, средняя и большая.
Признаки талассемии первой формы наименее тяжелые, хотя больные рискуют передать своим детям тяжелую форму заболевания.
Она встречается во всех уголках мира, но в основном локализуется в определенных географических районах. Особенно распространена в странах Средиземноморья и Среднего Востока. Около 20% населения этих районов — свыше 100 млн. человек — являются носителями гена. По месту локализации заболевание и получило свое название (производное от греческих слов «море» и «кровь»).
Анализ крови на талассемию
Обычно дети чувствуют себя хорошо и не выглядят больными. Зачастую Признаки талассемии обнаруживаются случайно при обычном анализе крови. У больных выявляется симптомы слабой анемии. Кровь под микроскопом напоминает кровь больных железодефицитной анемией, которая, безусловно, является наиболее распространенным типом анемии. Более тщательный анализ, однако, может исключить наличие дефицита железа и выявить аномальное количество нормальных компонентов крови.
Прогноз. Дети живут нормальной жизнью и не чувствуют себя больными. Однако в зрелости у них может развиться анемия, например в период беременности или при инфекционных заболеваниях.
Она развивается тогда, когда ребенок наследует дефектные гены, ответственные за производство бета-глобина, от обоих родителей. Его организм не вырабатывает нормальное количество бета-глобина. Избыточные альфа-глобины образуют нерастворимые сгустки в эритроцитах, делая клетки меньше и бледнее. Эти клетки живут меньше нормальных, кроме того, происходит снижение производства новых клеток В итоге развивается анемия в тяжелой форме. Однако очевидной она становится только в возрасте шести месяцев, так как в первые месяцы жизни большая часть гемоглобина — это фетальный, а не зрелый гемоглобин.
Дети выглядят беспокойными, бледными, у них плохой аппетит и малая прибавка в весе. Они поздно начинают ходить, но в умственном отношении развиваются нормально.
У ребенка появляется тяжелая анемия, которая сопровождается следующими симптомами:
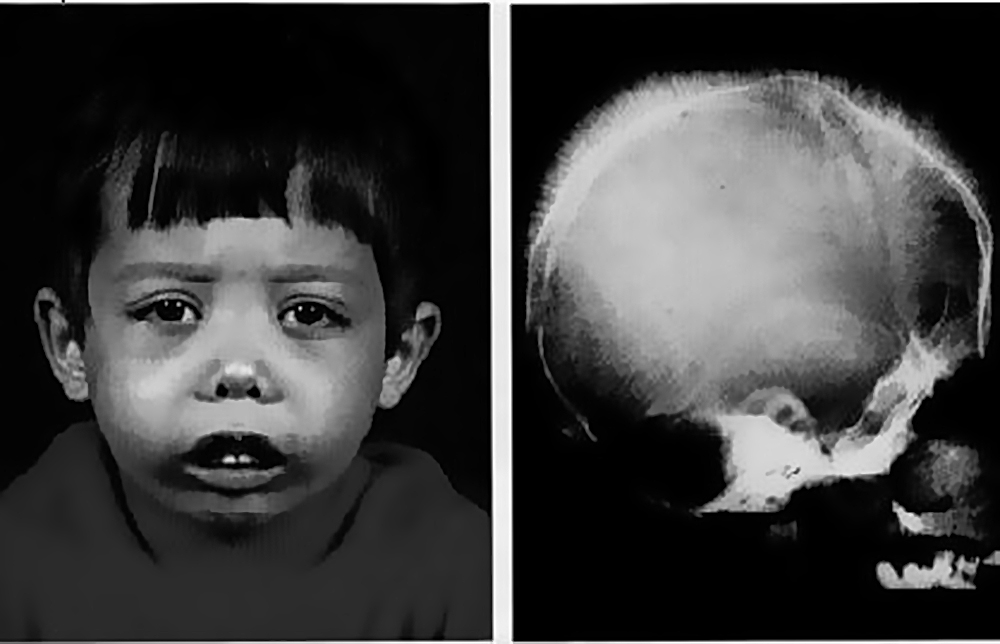
- характерное лицо, известное как монголоидное лицо,
- характерные изменения костей, видимые на рентгенограмме: череп будто встает «дыбом» вследствие разрастания костного мозга,
- истончение или переломы костей.
Кроме малой и большой бета талассемии существуют также средняя бета талассемия и альфа талассемия. Антенатальное исследование женщин с ее признаками помогает выявить плод с тяжелыми формами заболевания.
Третий тип называется так потому, что это более серьезное заболевание, чем малая, но менее серьезное, чем большая форма.
У пациентов с этой формой уровень гемоглобина относительно низок, но тем не менее достаточен, чтобы они могли вести приемлемый образ жизни. Чтобы провести лечение, им не требуются переливания крови, поэтому гораздо ниже риск проблем, связанных с избытком железа.
Прогноз лечения. Дети, у которых выявлены признаки этой формы талассемии развиваются нормально и часто доживают до совершеннолетия.
Она может принимать самые разные формы. Существует четыре набора генов, отвечающих за производство цепочек альфа-глобина, входящих в состав гемоглобина.
Степень тяжести заболевания зависит от того, сколько из этих четырех наборов генов поражено.
Если дефектен только одни набор, а три функционируют нормально, пациент не имеет никаких значительных болезненных проявлений. Но при двух и трех дефектных наборах заболевание постепенно принимает все более тяжелую форму. Тем не менее даже при трех дефектных наборах анемия часто слабая. При поражении всех четырех пасторов генов альфа-глобин не образуется. Это состояние несовместимо с жизнью, поэтому пораженный плод погибает еще до рождения. Лечение талассемии не проводится.
Это заболевание локализовано в районах распространения малярии. Она часто встречается в Юго-Восточной Азии, но отсутствует на Среднем Востоке и в Средиземноморье
Проводится по анализу крови. На ее наличие указывает анемия. В отличие от бета-талассемии, при этом заболевании в крови не наблюдается повышения уровня гемоглобина.
Теперь вы знаете основные признаки и способы лечения талассемии у детей. Здоровья вашему ребенку!
источник
Бета-Талассемии наиболее часто встречаются у выходцев с Индийского субконтинента, Средиземноморья и Ближнего Востока. В Великобритании дети, страдающие этими заболеваниями, рождаются от родителей индийского происхождения; в прошлом, многие были рождены от греков-киприотов, однако в настоящее время это происходит редко, поскольку в их сообществе проводится активное консультирование.
Поскольку дефицит синтеза b- и у-цепей продолжается после неонатального периода, производство увеличенной доли HbF и продукция 8-цепей увеличивают количество НbА2. Вследствие этого происходит преципитация цепей глобина внутри мембраны эритроцитов, приводящая к гибели клеток в костном мозге (неэффективный эритропоэз) и преждевременному удалению циркулирующих эритроцитов селезёнкой.
Тяжесть течения бета-талассемии зависит от количества присутствующих НbА и HbF.
• Большая бета-талассемия — НbА (а2р2) не может продуцироваться в связи с патологическим геном р-глобина. Наиболее тяжёлая форма.
• Умеренная бета-талассемия — мутации b-глобина позволяют образовываться небольшому количеству HbA и/или большому количеству HbR Более лёгкая, вариабельная.
• Малая бета-талассемия — один нормальный и один патологический гены b-глобина. Асимптомный носитель.
Большая бета-талассемия — аутосомно-рецессивное наследование мутаций в каждом из двух генов бета-глобина (по одному от каждого из родителей).
Клинические особенности талассемии у детей:
• Тяжёлая анемия и желтуха начиная с 3-6 мес жизни.
• Недостаточная прибавка в весе / задержка развития.
• Экстрамедуллярный гемопоэз, приводящий к распространению костного мозга, что обусловливает признак классического лица с гипертрофией верхней челюсти и выступающим лобным гребнем; отмечается значительная гепатоспленомегалия (не наблюдается у детей, которым проводилась адекватная трансфузия).
Это состояние в целом приводит к смерти без регулярных гемотрансфузий, поэтому всем пациентам проводятся пожизненные ежемесячные трансфузии эритроцитарной массы.
Целью является поддержание концентрации Hb выше 10 г/дл для того, чтобы уменьшить отставание в развитии и предотвратить деформацию костей. Повторные гемотрансфузий приводят к хронической перегрузке железом, которая вызывает сердечную недостаточность, цирроз печени, диабет, бесплодие и отставание в развитии. Для того чтобы минимизировать этот риск, терапия хелатами железа с помощью подкожных инъекций дезферриоксамина, проводимая в ночь перед трансфузией, начинается с 2-3-летнего возраста.
Пациенты, которые хорошо отвечают на трасфузию и хелацию, имеют 90% шанс на то, что они доживут до сорока лет и старше. Однако сложно достигнуть комплайнса. Те, которые не могут переносить лечение, имеют высокую смертность в раннем детстве вследствие перегрузки железом. Альтернативным лечением большой бета-талассемии является трансплантация костного мозга, которая в настоящее время является единственным способом излечения.
Она обычно оставляется в резерве для детей, имеющих братьев или сестёр, идентичных по HLA (главный комплекс гистосовместимости), поскольку в таком случае имеется 90% шансов на успех (т.е. независимость от трансфузий и долгосрочное излечение), и в то же время 5% шансов смерти в связи с трансплантацией.
Осложнения долговременной трансфузии у детей при бета-талассемии:
• Отложение железа — наиболее важное (у всех пациентов):
— Сердце — кардиомиопатия.
— Печень — цирроз.
— Поджелудочная железа — диабет.
— Гипофиз — задержка развития и сексуального созревания.
— Кожа — гиперпигментация.
• Образование антител (10% детей):
— Алло-антитела у пациента сильно осложняют подбор совместимой группы крови.
• Инфекции — в настоящее время нетипичны ( 5,5х1012/л). Наиболее важной диагностической особенностью является увеличение НbА2 (и приблизительно в половине случаев отмечается умеренное увеличение уровня HbF на 1-3%) на электрофорезе Нb.
Лёгкая бета-талассемия может быть спутана с железодефицитной анемией лёгкой степени, что приводит к ненужной терапии железом.
У здоровых индивидуумов имеются четыре гена альфа-глобина. Проявления синдромов а-талассемии зависят от количества функционирующих генов этого белка.
Наиболее тяжёлая а-талассемия, большая альфа-талассемия (также известная как Нb Барте), возникает в результате делеции всех четырёх генов альфа-глобина, поэтому не может производиться никакого HbA (a2p2). Она обычно встречается в семьях родом из Юго-Западной Азии и проявляется в среднем триместре в форме водянки плода (отёк и асцит) вследствие анемии плода, которая всегда заканчивается гибелью во внутриутробном периоде или в течение нескольких часов после рождения.
Долгосрочное выживание при большой а-талассемии отмечается лишь у тех, кому проводились ежемесячные внутриутробные трансфузии до рождения с последующими пожизненными ежемесячными трансфузиями после рождения. Диагноз устанавливается с помощью электрофореза Нb или высокоэффективной жидкостной хроматографии (ВЭЖХ), на которой выявляют только Нb Барте.
Если происходит делеция только трёх генов а-глобина (болезнь НbН), у детей с таким состоянием отмечается лёгкая/умеренная анемия, но некоторые пациенты нуждаются в трансфузии.
Делеция одного или двух генов а-глобина (известная как лёгкая альфа-талассемия) обычно бессимптомная и анемия лёгкая или отсутствует. Могут быть гипохромия и микроцитарность эритроцитов, поэтому её можно спутать с железодефицитным состоянием.
источник
Талассемия – наследственные гемоглобинопатии, характеризующиеся угнетением синтеза цепочечных белковых молекул, образующих структуру гемоглобина. Это приводит к повреждению мембраны эритроцитов и разрушению красных клеток крови с развитием гемолитических кризов. Признаками талассемии служат характерные костные изменения, гепатоспленомегалия, анемический синдром. Диагноз талассемии подтверждается клиническими и лабораторными данными (исследованием гемограммы, гемоглобина, миелограммы, электрофоретическим методом). Возможна пренатальная диагностика талассемии. В лечении талассемии применяются гемотрансфузии, терапия десфералом, спленэктомия, трансплантация костного мозга.
Талассемия – группа генетически детерминированных болезней крови, развивающихся при нарушении синтеза a- или β-цепей гемоглобина, сопровождающихся гемолизом, гипохромной анемией, микроцитозом. В гематологии талассемия относится к наследственным гемолитическим анемиям — количественным гемоглобинопатиям. Талассемия широко распространена среди населения Средиземноморского и Черноморского региона; название заболевания буквально переводится как «анемия морского побережья». Также случаи талассемии нередки в странах Африки, Ближнего Востока, Индии и Индонезии, Средней Азии и Закавказья. С синдромом талассемии каждый год в мире рождается 300 тыс. детей. В зависимости от формы патологии течение талассемии может быть тяжелым, фатальным или легким, бессимптомным. Так же, как серповидно-клеточная анемия, талассемия играет роль защитного фактора против малярии.
С учетом поражения той или иной полипептидной цепи гемоглобина различают:
- a-талассемию (с подавлением синтеза альфа-цепей HbA). Данная форма может быть представлена гетерозиготным носительством манифестного (α-th1) или немого (α-th2) гена; гомозиготной a-талассемией (водянкой плода с гемоглобином Бартса); гемоглобинопатией Н
- b-талассемию (с подавлением синтеза бета-цепей HbA). Включает в себя гетерозиготную и гомозиготную β-талассемию (анемию Кули), гетерозиготную и гомозиготную δβ-талассемию (F-талассемию)
- γ-талассемию (с подавлением синтеза гамма-цепей гемоглобина)
- δ-талассемию (с подавлением синтеза дельта-цепей гемоглобина)
- талассемию, обусловленную нарушением структуры гемоглобина.
Статистически чаще встречается β-талассемия, которая, в свою очередь, может протекать в 3-х клинические формах: малой, большой и промежуточной. По тяжести синдрома выделяют легкую форму талассемии (пациенты доживают до половой зрелости), средне-тяжелую (продолжительность жизни больных 8-10 лет) и тяжелую (гибель детей наступает в первые 2-3 года жизни).
Талассемия является генетическим заболеванием с аутосомно-рецессивным наследованием. Непосредственной причиной патологии выступают различные мутационные нарушения в гене, кодирующем синтез той или иной цепи гемоглобина. Молекулярную основу дефекта могут составлять синтез аномальной матричной РНК, делеции структурных генов, мутации регуляторных генов либо их неэффективная транскрипция. Следствием подобных нарушений служит снижение или отсутствие синтеза одной из полипептидных гемоглобиновых цепей.
Так, при b-талассемии бета-цепи синтезируются в недостаточном количестве, что приводит к избытку альфа-цепей, и наоборот. Избыточно продуцируемые полипептидные цепи откладываются в клетках эритроидного ряда, вызывая их повреждение. Это сопровождается деструкцией эритрокариоцитов в костном мозге, гемолизом эритроцитов в периферической крови, гибелью ретикулоцитов в селезенке. Кроме этого, при b-талассемии в эритроцитах накапливается фетальный гемоглобин (НbF), не способный транспортировать кислород в ткани, что вызывает развитие тканевой гипоксии. Вследствие костномозговой гиперплазии развивается деформация костей скелета. Анемия, тканевая гипоксия и неэффективный эритропоэз в той или иной степени нарушают развитие и рост ребенка.
Для гомозиготной формы талассемии характерно наличие двух дефектных генов, унаследованных от обоих родителей. При гетерозиготном варианте талассемии пациент является носителем мутантного гена, унаследованного от одного из родителей.
Признаки большой (гомозиготной) b-талассемии проявляются уже в течение 1-2-го года жизни ребенка. Больные дети имеют характерное монголоидное лицо, седловидную переносицу, башенный (четырехугольный) череп, гипертрофию верхней челюсти, нарушение прикуса, гепато- и спленомегалию. Проявлениями анемизации служат бледный или землисто-желтушный цвет кожных покровов. Поражение трубчатых костей сопровождается отставанием в росте и патологическими переломами. Возможно развитие синовита крупных суставов, калькулезного холецистита, язв нижних конечностей. Фактором, осложняющим течение b-талассемии, выступает гемосидероз внутренних органов, приводящий к развитию цирроза печени, фиброза поджелудочной железы и, как следствие, — сахарного диабета; кардиосклероза и сердечной недостаточности. Больные восприимчивы к инфекционным заболеваниям (кишечным инфекциям, ОРВИ и др.), возможно развитие тяжелых форм пневмонии и сепсиса.
Малая (гетерозиготная) b-талассемия может протекать бессимптомно или с минимальными клиническими проявлениями (умеренным увеличением селезенки, незначительно выраженной гипохромной анемией, жалобами на повышенную утомляемость). Аналогичная симптоматика сопровождает течение гетерозиготной формы a-талассемии.
При гомозиготной форме a-талассемии альфа-цепи полностью отсутствуют; фетальный гемоглобин у плода не синтезируется. Данная форма талассемии несовместима с жизнью, что приводит к внутриутробной гибели плода вследствие развивающегося синдрома водянки или самопроизвольному прерыванию беременности. Течение гемоглобинопатии Н характеризуется развитием гемолитической анемии, спленомегалии, тяжелых костных изменений.
Талассемию следует заподозрить у лиц с семейным анамнезом, характерными клиническими признаками и лабораторными показателями. Больные талассемией нуждаются в консультации гематолога и медицинского генетика.
Типичными гематологическими изменениями служат снижение уровня гемоглобина и цветового показателя, гипохромия, наличие мишеневидных эритроцитов, повышение уровня железа сыворотки крови и непрямого билирубина. Электрофорез Hb на ацетат-целлюлозной пленке используется для определения различных гемоглобиновых фракций. При изучении пунктата костного мозга обращает внимание гиперплазия красного кроветворного ростка с высоким числом эритробластов и нормобластов. Молекулярно-генетические исследования позволяют выявить мутацию в локусе a- или β-глобина, нарушающую синтез полипептидной цепи.
На краниограммах при большой b-талассемии выявляется игольчатый периостоз (феномен «волосатого черепа»). Характерна поперечная исчерченность трубчатых и плоских костей, наличие мелких очагов остеопороза. С помощью УЗИ брюшной полости обнаруживается гепатоспленомегалия, камни желчного пузыря.
При подозрении на талассемию требуется исключить железодефицитную анемию, наследственный микросфероцитоз, серповидно-клеточную анемию, аутоиммунную гемолитическую анемию. В семьях, имеющих больных талассемией, рекомендуется проведение генетического консультирования супругов и инвазивной дородовой диагностики (биопсии хориона, кордоцентеза, амниоцентеза) для выявления гемоглобинопатии на ранних сроках беременности. Подтверждение гомозиготных форм талассемии у плода служит показанием для искусственного прерывания беременности.
Лечебная тактика при различных формах талассемии неодинакова. Так, пациенты с малой b-талассемией в лечении не нуждаются. С другой стороны, больным с гомозиготной b-талассемией с первых месяцев жизни требуется проведение гемотрансфузионной терапии (переливание размороженных или отмытых эритроцитов), введение хелатирующих препаратов, связывающих железо (дефероксамина), глюкокортикоидов при возникновении гемолитических кризов. При всех формах талассемии показан прием препаратов фолиевой кислоты и витаминов группы В.
При гиперспленизме (особенно на фоне гемоглобиноза Н) требуется удаление селезенки (спленэктомия). Из-за склонности к присоединению инфекционных осложнений больным рекомендуется обязательная вакцинация против пневмококковой инфекции. Многообещающим методом лечения талассемии служит трансплантация костного мозга от гистосовместимого донора.
Прогноз больших форм талассемии неблагоприятный; больные погибают в младенческом или молодом возрасте. При гетерозиготной бессимптомной форме талассемии продолжительность и качество жизни в большинстве случаев не страдают. Первичная профилактика талассемии включает предупреждение браков между гетерозиготными носителями генов заболевания, а при высоком генетическом риске рождения больного потомства – отказ от деторождения.
источник
Талассемия (анемия Кули) — это группа наследственных расстройств, которые влияют на количество гемоглобина, производимых человеком. Гемоглобин относится к семейству соединений, состоящих из гема (железосодержащий комплекс) и различных глобинов (белковые цепи, которые окружают гемовый комплекс).
Молекулы гемоглобина (Hb) находятся во всех эритроцитах и являются причиной их красного цвета. Они связывают кислород в легких, переносят его через кровоток и выделяют в ткани организма. Различные типы гемоглобина классифицируются в соответствии с типом белковых цепей, которые они содержат.
Нормальные гемоглобины взрослых включают в себя:
- Гемоглобин А (составляет около 95% – 98% взрослого Hb). HbA содержит две альфа (α) белковые цепи и две бета (ß) цепи.
- HbA2 (составляет около 2% – 3,5% взрослого Hb), имеет две альфа (α) и две дельта (δ) цепи.
- HbF (до 2%). Это основной гемоглобин, вырабатываемый плодом во время беременности. Его производство обычно падает до низкого уровня в течение года после рождения. HbF имеет две альфа (α) и две гамма (γ) цепи.
Мутации в генах, кодирующих глобиновые цепи, могут вызывать нарушения в выработке гемоглобина. Есть 4 гена, которые кодируют цепи альфа-глобина, и 2 гена, которые кодируют цепи бета-глобина.
Наследственные нарушения производства гемоглобина делятся на две категории:
- Талассемия: снижение выработки нормальных гемоглобинов
- Гемоглобинопатия: производство аномальной молекулы гемоглобина
Талассемии представляют собой группу нарушений, при которых мутации в одном или нескольких из генов альфа- или бета-глобина вызывают уменьшение количества продуцируемого HbA. Это приводит к снижению уровня HbA, относительному увеличению количества минорных гемоглобинов HbA2 и HbF и, возможно, обнаружению необычных типов гемоглобина.
Талассемии обычно классифицируются по типу глобиновой цепи, синтез которой снижается.
Альфа-талассемия возникает вследствие делеции или мутации в одной, или нескольких копиях гена 4-альфа-глобина. Чем больше затронутых генов, тем меньше вырабатывается альфа-глобина. Четыре различных типа альфа-талассемии включают в себя:
- Черта альфа-талассемии (1 пораженный ген). Безмолвный носитель будет иметь нормальные уровни гемоглобина и индексы эритроцитов, которые являются нормальными или демонстрируют слегка уменьшенный MCH (гипохромия). Носители могут передать пораженный ген своим потомкам. Часто эти люди идентифицируются только после рождения ребенка с болезнью HbH или признаком альфа-талассемии.
- Черта альфа-талассемии (2 пораженных гена). Пациенты с признаком альфа-талассемии имеют меньшие (микроцитарную), бледные (гипохромную) эритроциты и легкую хроническую анемию, но обычно они не испытывают никаких симптомов. Это анемия, которая не реагирует на добавки железа. Диагностика признака альфа-талассемии обычно происходит путем исключения других причин микроцитарной анемии. Подтверждающее тестирование с помощью анализа ДНК является важной частью процесса диагностики альфа-талассемии, поскольку на каждой хромосоме имеется две копии (HbA1 и HbA2) гена альфа-цепи. Если человек потерял обе копии из одной хромосомы, это имеет различные последствия для передачи более серьезной формы заболевания своим детям, чем если бы они потеряли одну копию из каждой двух разных хромосом. Смотрите раздел об анализе ДНК ниже.
- Болезнь гемоглобина Н (3 пораженных гена). При этом условии большое уменьшение количества производимых цепей альфа-глобина вызывает избыток бета-цепей, которые затем агрегируют в тетрамеры бета4 (группы из 4 бета-цепей), известные как гемоглобин H. Болезнь HbH может вызывать анемию от средней до тяжелой степени и спленомегалию (увеличенная селезенка). Клиническая картина, связанная с заболеванием HbH, чрезвычайно разнообразна. У некоторых людей она протекает бессимптомно, в то время как другие имеют тяжелую анемию. Болезнь гемоглобина Н чаще всего встречается у лиц юго-восточного или восточно-средиземноморского происхождения.
- Большая альфа-талассемия (также называемая фетальный гидропс, 4 пораженных гена). Это самая тяжелая форма альфа-талассемии. В этих условиях альфа-глобин не вырабатывается, следовательно, HbA или HbF также не вырабатываются. Плоды, пораженные альфа-талассемией, становятся анемичными на ранних сроках беременности. Они становятся отечными (сохраняют жидкость), и часто имеют увеличенные сердца и печень. Этот диагноз часто ставится в последние месяцы беременности, когда УЗИ плода указывает на отек плода. Приблизительно в 80% случаев у матери возникает токсемия и может развиться тяжелое послеродовое кровотечение. Дети с большой альфа-талассемией обычно недоношенные, мертворожденные или умирают вскоре после рождения.
Люди с альфа-талассемией могут быть неправильно диагностированы неосторожными врачами как дефицит железа, так как дефицит железа также приводит к появлению небольших бледных (микроцитарных гипохромных) эритроцитов.
Важно, чтобы лечение железом у больных талассемией назначалось только тогда, когда специфические анализы на железо (ферритин, сывороточное железо, TIBC, трансферрин) подтвердили дефицит железа. Это особенно важно при альфа-талассемии, где существует небольшая вероятность развития опасной перегрузки железом.
Альфа-талассемия чаще всего встречается у людей с этническим происхождением из Юго-Восточной Азии, Южного Китая, Ближнего Востока, Индии, Африки и Средиземноморья.
Бета-талассемия происходит из-за мутаций в одном или обоих генах бета-глобина. Было выявлено от 100 до 200 мутаций, но распространены только около 20.
Тяжесть анемии, вызванной бета-талассемией, зависит от того, какие мутации присутствуют, и от того, уменьшают ли они выработку бета-глобина (так называемая бета + талассемия) или полностью ее устраняют (так называемая бета-талассемия). Различные типы бета-талассемии следующие:
- Бета-талассемия с характерной чертой. У человека с этим заболеванием один нормальный ген и один с мутацией. Обычно они не испытывают никаких проблем со здоровьем, кроме микроцитоза (маленьких эритроцитов) и возможной легкой анемии, которая не будет реагировать на добавки железа. Эта генная мутация может передаваться детям индивидуума.
- Талассемия интермедиа. В этом состоянии у больного человека есть два ненормальных гена, но он все еще производит некоторое количество бета-глобина. Тяжесть возникшей анемии и проблемы со здоровьем зависят от имеющихся мутаций. Разграничительной линией между промежуточной талассемией и большой талассемией является степень анемии, а также количество и частота переливаний крови, необходимых для ее лечения. Тем, у кого промежуточная талассемия, иногда требуется переливание, но не на регулярной основе.
- Талассемия крупная. Это самая тяжелая форма бета-талассемии. У пациента есть два ненормальных гена, которые вызывают либо серьезное снижение, либо полное отсутствие продукции бета-глобина, предотвращая выработку значительных количеств HbA. Это состояние обычно появляется у ребенка после 3-месячного возраста и вызывает опасную для жизни анемию. Эта анемия требует регулярных переливаний крови в течение всей жизни и значительной постоянной медицинской помощи. Со временем эти частые переливания приводят к чрезмерному количеству железа в организме. При отсутствии лечения это избыточное железо может откладываться в печени, сердце и других органах и может привести к преждевременной смерти от недостаточности органов.
Другие формы талассемии возникают, когда ген бета-талассемии наследуется в сочетании с геном варианта гемоглобина. Наиболее важными из них являются:
- HbE – бета-талассемия. HbE является одним из наиболее распространенных вариантов гемоглобина, встречающихся преимущественно у людей юго-восточного азиатского происхождения. Если человек наследует один ген HbE и один ген бета-талассемии, комбинация провоцирует талассемию HbE-бета, которая вызывает умеренно тяжелую анемию, подобную промежуточной бета-талассемии.
- HbS – бета-талассемия или серповидноклеточная – бета-талассемия. HbS является одним из наиболее известных вариантов гемоглобина. Наследование одного гена HbS и одного гена бета-талассемии приводит к талассемии HbS-бета. Тяжесть состояния зависит от количества бета-глобина, продуцируемого бета-геном. Если бета-глобин не вырабатывается, клиническая картина практически идентична серповидноклеточной анемии.
Общий клинический анализ крови (ОАК). ОАК — это анализ клеток и жидкости в крови. Помимо прочего, ОАК сообщит врачу, сколько эритроцитов в крови, сколько гемоглобина содержится в них, и поможет измерить размер и форму присутствующих эритроцитов. Эти переменные называются индексами эритроцитов и включают MCH (средний корпускулярный гемоглобин) и MCV (средний корпускулярный объем), как измерения содержания гемоглобина и размера эритроцитов.
Низкий MCH или MCV часто являются первым признаком талассемии. Если уровень MCH или MCV низкий, а дефицит железа исключен, человек может быть носителем талассемии. При дефиците железа трудно исключить талассемию.
Микроскопическое исследование мазка крови. При этом тесте тонкий микроскопический слой крови исследуют на предметном стекле под микроскопом. Количество и тип лейкоцитов, эритроцитов и тромбоцитов можно подсчитать вручную и оценить, являются ли они нормальными и зрелыми. Различные нарушения влияют на нормальное производство клеток крови. При талассемии эритроциты часто бывают микроцитарными (маленькими) с низким MCV. Они также могут:
- Быть гипохромным (бледный — указывает низкий гемоглобин, чем обычно).
- Варьироваться по размеру (анизоцитоз) и форме (пойкилоцитоз).
- Быть зародышем (не нормально в зрелом эритроците).
- Быть искаженными, выглядеть под микроскопом как яблочки.
Анализ железа. Анализ может включать: железо, ферритин, UIBC, TIBC и процентное насыщение трансферрина. Эти тесты измеряют различные аспекты хранения и использования железа в организме. Они помогают определить, является ли дефицит железа причиной и/или обострением анемии пациента. Одному или нескольким пациентам с талассемией также может быть назначено наблюдение за степенью перегрузки железом.
Оценка гемоглобинопатии (Hb). Этот тест измеряет тип и относительное количество гемоглобинов, присутствующих в эритроцитах. Гемоглобин А, состоящий из альфа- и бета-глобина, является основным нормальным типом гемоглобина, обнаруживаемым у взрослых. Больший процент HbA2 и/или HbF обычно наблюдается при признаке бета-талассемии. HbH может наблюдаться при альфа-талассемии, но только тогда, когда по меньшей мере два из четырех альфа-генов удалены или мутированы.
Анализ ДНК, Во многих случаях анализ ДНК не требуется, поскольку диагноз может быть поставлен по результатам вышеуказанных тестов. Анализ ДНК чаще всего используется в семьях, страдающих альфа-талассемией. Этот тест используется для изучения делеций и мутаций в альфа и реже генах, продуцирующих бета глобин.
Семейные исследования могут быть проведены, чтобы оценить статус носителя и типы мутаций, присутствующих в других членах семьи. Исследование ДНК является важным инструментом для установления точного диагноза талассемии. В частности, при альфа-талассемии важно знать, имеет ли человек с чертой альфа-талассемии два мутированных гена в одной хромосоме или по одному в каждой хромосоме.
Большинство людей с талассемией не нуждаются в лечении. Всем людям с диагнозом талассемия, которые планируют семью, настоятельно рекомендуется обратиться за генетическим советом, чтобы понять последствия для своего потомства. Требуется, чтобы лаборатория провела генетическое исследование партнера, чтобы генетический консультант предоставил точную информацию о риске передачи затронутых генов своим детям и серьезности талассаземии или гемоглобинопатии, которая может развиться.
Пациенты с гемоглобином Н или промежуточной бета-талассемией будут испытывать различные степени анемии в течение всей жизни. Они могут вести относительно нормальную жизнь, но требуют регулярного мониторинга и иногда могут нуждаться в переливании крови.
Добавки с фолиевой кислотой часто назначаются для борьбы с анемией, но следует избегать приема препаратов железа, если дефицит железа не подтвержден более конкретными исследованиями.
Часто изменения в показателях эритроцитов наблюдаются, когда женщина беременна, это может иногда приводить к тяжелой анемии. Понимание влияния диагноза талассемии на беременность и возможных последствий для здоровья потомства пары является одной из наиболее важных причин для установления точного диагноза талассемии.
Больным с бета-талассемией обычно требуется переливание крови каждые 3-4 недели в течение всей жизни. Эти переливания помогают поддерживать гемоглобин в достаточно высокой концентрации, чтобы обеспечить организм кислородом и предотвратить нарушения роста и повреждения органов.
Частые переливания, однако, поднимают железо до токсичных уровней, что приводит к отложению железа в печени, сердце и других органах.
Регулярная хелатная терапия железом используется для снижения уровня железа в организме Это включает введение препарата, который связывается с железом и помогает вымывать его из организма через мочу. Также часто больным может требоваться спленэктомия (хирургическая операция по удалению селезёнки).
Дети с большой альфа-талассемией обычно рождаются недоношенными, мертворожденные или умирают вскоре после рождения. Лечение сосредоточено на выявлении состояния и прерывании беременности или наблюдении за осложнениями у матери.
Экспериментальные методы лечения, такие как переливание крови плода, были успешными в очень немногих случаях при рождении ребенка.
источник
Наследственность играет большую роль в развитии патологических состояний человека. Ярким примером ее негативного влияния выступает гемоглобинопатия талассемия. Это собирательное понятие группы болезней системы крови, возникающих вследствие нарушения образования гемоглобина в красных кровяных тельцах.
Благодаря врачу Дж.Уипплу, который заметил увеличение случаев поражения людей средиземноморской расы неизвестной «хворью», возникло название болезни, что означало с греческого «таласса» — море и «эмиа» — кровь. Американский педиатр Т.Б.Кули видел много пораженных детей и был озадачен проблемой – талассемия что это — новый вирус, бактерия? В 1925 году Кули, благодаря его длительным исследованиям и усердной работе, подробно описал болезнь.
Причины талассемии сводятся к унаследованию от родителей по аутосомно-рецессивному типу мутагенного гена. Клинические проявления будут в случае наследования поврежденного гена от обоих родителей. Если «сбой» только у одного из родителей, риск болезни снижается к 25-50%, или же не заболеет вовсе. Мутация генов происходит под влиянием факторов, которые прямо или опосредованно влияют на организм человека.
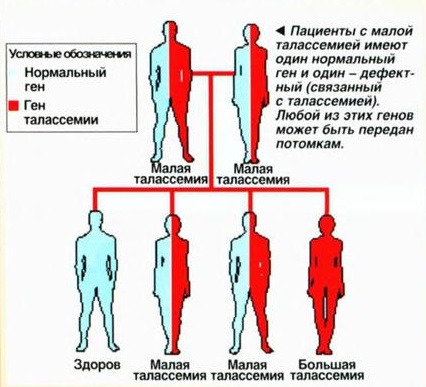 Схема наследования талассемии
Схема наследования талассемии
К ним относятся:
- Химические соединения табачного дыма (бензолы, фенолы) действуют мутагенно на генном уровне
- Алкоголь, а именно содержащийся в нём этанол, угнетает резистентность организма к воздействию повреждающих факторов.
- Перенесенные вирусные заболевания (грипп, корь, краснуха, герпетическая инфекция и др). Частицы вирусной инфекции представляют собой ДНК, внедряясь в гены клеток, они способствуют их изменению и клетки синтезируют себе подобных клонов-мутантов.
- Ионизирующее излучение и радиация окружает человека повсеместно: микроволновые печи, современные гаджеты, компьютеры, рентгеновские обследования, радиотерапия в окологии, последствия Чернобыльской катастрофы. Радиационные потоки пронизывают тело человека, оставляя следы на генетическом аппарате.
- Мутагены химических веществ (свинец, ртуть, пестициды, нитраты, двуокись серы), чему подвержены работники промышленности.
- Медицинские препараты (цитостатики, метилксантины, антиконвульсанты, прихотропные, гормоны).
Гемоглобин состоит из гема и белка глобина. В каждой молекуле 4 цепи — 2 альфа и 2 бета, поражение одной из полипептидных цепей и определяет развитие талассемии. В результате возникает нехватка кислорода, провоцирующая усиленное выделение эритропоэтина, что в свою очередь активирует увеличение образования в костном мозге новых эритроцитов в крови с его гиперплазией и последующей деформацией костной системы.
Гемоглобин группируют на:
- HbU – эмбриональный, функционирующий внутриутробно с третьей по десятую неделю;
- HbF – фетальный, основной гемоглобин плода, у новорожденных составляет 18-20%;
- HbA – взрослый, с двухлетнего возраста и до конца жизни на уровне 98%;
- HbA2 – малый компонент взрослого.
В зависимости от локализации мутации выделяют талассемию А, В и АВ.
Талассемия АВ несовместимая с жизнью.
А-талассемия характеризуется дефектом генов в 16 паре хромосом с нарушением синтеза альфа цепей глобина. Бета цепи функционируют в полном объёме, но они не способны в нужном количестве распределять кислород.
По количества мутагенных генов талассемия А делится на:
- Бессимптомную (поражение одного гена, клинические проявления отсутствует, но у такого человека есть минимальная угроза рождения ребенка с талассемией);
- Малую (мутации в двух генах из четырёх, оставшиеся могут обеспечивать организм газами в необходимом количестве, минимальные проявления симптомов с риском болезни у детей 25%);
- Н-гемоглобинопатию (три гена не работают, обеспечение кислородом на критическом уровне, угрожающая жизни форма при несвоевременном обращении за лечением);
- Истинную, или водянку мозга, плода (тотальное поражение, гибель плода внутриутробно, очень редко после рождения, в этом случае смерть через несколько минут).
В-талассемия наиболее часто встречается. Патогенез возникновения аналогичный, но поврежденный ген локализован в 11 паре хромосом, с уменьшением количества синтеза бета цепей. В результате формируется комплекс из цепей альфа, подверженный быстрому распаду и впоследствии гибели эритроцитов.
Классифицируют по принципу количества мутагенных генов:
- Большая талассемия – характеризуется повреждением двух генов, протекает с жизненно опасными нарушениями в виде склерозирующих изменений печени и поджелудочной железы, некрозов кожи, ангиопатиями нижних конечностей.
- Промежуточная талассемия – мутации одного или двух генов, но со способностью к выработке необходимого гемоглобина. Симптоматика менее выражена, в первуюочередь страдает костная система и органы кровотворения.
- Малая талассемия – выпадение из работы одной цепи, без влияния на состояние больного. В редких случаях протекает с анемией.
Степени тяжести по выраженности клинических проявлений:
- Тяжелая — обнаруживается в первые минуты после рождения с риском смерти;
- Средней тяжести — на поддерживающей терапии дети доживают до 8-12 лет;
- Лёгкая – дети ничем не отличаются от ровесников, первые симптомы в зрелом возрасте.
При тяжелых и умеренных формах, талассемия крови у детей выявляется в первые дни после рождения. При легком течении заболевание обнаруживают в зрелом возрасте, или оно остается не распознанным. Симптомы талассемии выделены в 4 основные группы в соответствии с патогенетическим изменениям.
Клиника нарушения образования красных клеток:
- бледная, тусклая, сухая кожа;
- низкий рост и недостаточный вес соответственно центильным таблицам, из-за замедления процессов митоза;
- снижение/отсутствие аппетита;
- хроническая усталость, отсутствие интереса к обучению, умственная отсталость, снижение концентрации, раздражительность, апатия вследствие кислородного голодания;
- изъязвления кожи верхних и нижних конечностей при тяжелых формах с сильной гипоксией;
- гепатоспленомегалия как компенсаторная реакция, сопровождается тяжестью в подреберье, диспепсией.
Симптомы, связанные с гиперплазией костного мозга:
- сплющенная переносица;
- увеличение головы, несоответствующее возрасту;
- уменьшение глазных щелей;
- чрезмерное увеличение размеров верхней челюсти;
- переломы костей из-за изменения их структуры.
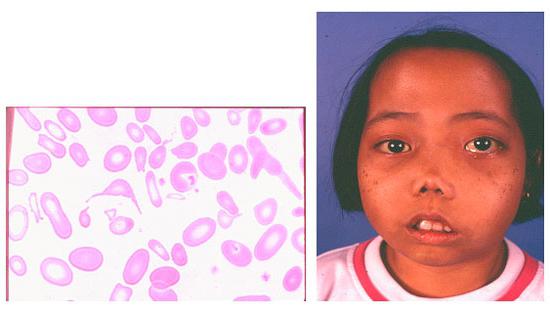 Проявление талассемии
Проявление талассемии
Вследствие нарушения выведения билирубина:
- желтый цвет кожи за счет гипербилирубинемии. В нормальных условиях при завершении жизни эритроцита, гемоглобин в крови путём ряда реакции преобразовывается в желчный пигмент билирубин, который выводится печенью. При талассемии крови количество билирубина патологически увеличивается и возможности печени не способны в полном объёме вывести его. Пожелтение кожи и слизистых говорят о неблагоприятном прогнозе;
- также, в результате его накопления образуются билирубиновые камни в желчном пузыре;
Вследствие накопления мочевой кислоты и уратов:
- Ограничение движений и боли в суставах;
- Камни в почках, с клиникой почечной колики.
- Стеноз почечных артерий;
Симптомы гемохроматоза, возникающего после многократных процедур по переливанию крови:
- специфическая гиперпигментация кожи темно-коричневого цвета;
- замедление роста и позднее половое созревание как результат накопления железа в гипофизе;
- тахи- или брадикардия сердца, признаки сердечной недостаточности, инфаркт миокарда;
- разрушение клеток печени с замещение соединительной тканью и развитием цирроза или рака;
- хроническая почечная недостаточность с необходимостью пересадки почки или пожизненная необходимость в аппарате искусственной почки.
К основным методам диагностики относят:
- развернутый анализ крови и мазок на предметное стекло;
- биохимический анализ крови (уровень билирубина, железа, мочевой кислоты, аланинаминотрансферазы и аспартатаминотрансферазы);
- ПЦР.
Дополнительные:
- рентгенография ОГК, ОБП и головы;
- ультразвуковое исследование;
- пункция костного мозга.
- ОЖСС, ферритин в крови.
 УЗИ головы
УЗИ головы
Диагностика талассемии должна проводится в условиях стационара в предельно ранние сроки после появления первых признаков болезни.
Анализ крови – многоинформативный, доступный метод, который выполняют все лаборатории.
Таблица главных показателей, которые изменяются при талассемии:
| Значения нормы | При талассемии |
| Эритроциты Помимо этого, в мазке под микроскопом обнаруживается изменение размеров эритроцитарных клеток в виде микроцитоза — уменьшение диаметра до 5-6 мкм, при норме 7,5-8 мкм. Для талассемии характерны мишеневидные эритроциты, светлые по периферии и темные в центре ( в норме равномерно красные).
Уровень общего билирубина крови в пределах нормы или повышен (норма 20,4 мкмоль/литр). Непрямой билирубин в крови — резко повышен до 100-200 мкм/л и выше, при норме до 17 мкмоль/л. АлАТ, АсАТ — показатели поражения печени, сердечной мышцы, значительно увеличиваются, свидетельствуют о тяжести процесса. Мочевая кислота – (N до 8,0 ммоль/л). Увеличивается при развитии гепатоспленомегалии до высоких цифр . Сывороточное железо (N у женщин 14,5-17,7 мкмоль/л, у мужчин 18-22,5 ммоль/л) – повышено. Пропорционально ему одновременно растет количество ферритина сыворотки крови.
Материал для исследования – кровь, моча, слюна. Суть метода в выявлении поврежденного гена и хромосомы.
Печень и селезенка увеличены, неоднородной эхогенности, участки склероза ткани печени. В почках и лоханке – уратные камни.
Несмотря на высокие достижения в отрасли медицины, лекарства для излечения больного талассемией не существует. Лечение талассемии направленно на улучшение общего состояния и поддержания его на стабильном уровне, предотвращения жизненно опасные осложнения. Главные направления оказания помощи:
В отношении жизни — благоприятный при успешной пересадке костного мозга. Относительно благоприятный при минимальной форме талассемии, легком течении. Неблагоприятный при талассемии майор. В отношении выздоровления – неблагоприятный, исключение составляет пересадка с положительной динамикой. Отказ от курения и алкоголя, правильное питание, применение средств защиты на производстве, обязательное обследование беременных, если родители носители болезни. источник |
 Переливание крови
Переливание крови